Biochemistry
Amino acids, protein structure, enzyme kinetics, carbohydrate metabolism, lipid metabolism, nucleotide metabolism, vitamins, cofactors, and every metabolic pathway, enzyme deficiency, and clinical correlation across the full scope of medical biochemistry.
01 Overview & Scope of Medical Biochemistry
Biochemistry is the study of chemical processes within and relating to living organisms. In medical education, it serves as the molecular foundation for understanding physiology, pharmacology, pathology, and genetics. Virtually every clinical discipline relies on biochemical principles—from diabetic ketoacidosis in the emergency department to enzyme replacement therapy for lysosomal storage diseases, from pharmacokinetics in anesthesia to tumor metabolism in oncology.
A solid command of biochemistry is essential for interpreting lab values, understanding drug mechanisms, diagnosing inborn errors of metabolism, and reasoning through the metabolic derangements encountered in critical care, endocrinology, and nearly every other clinical specialty.
Core Domains of Medical Biochemistry
| Domain | Scope | Clinical Relevance |
|---|---|---|
| Amino acids & proteins | Structure, folding, hemoglobin, collagen | Sickle cell disease, osteogenesis imperfecta, Ehlers-Danlos |
| Enzyme kinetics | Michaelis-Menten, inhibition, regulation | Drug design (competitive vs. non-competitive inhibitors), enzymopathies |
| Carbohydrate metabolism | Glycolysis, TCA, ETC, gluconeogenesis, glycogen | Diabetes, glycogen storage diseases, lactic acidosis |
| Lipid metabolism | Beta-oxidation, ketogenesis, lipoproteins, cholesterol | Familial hypercholesterolemia, DKA, fatty acid oxidation defects |
| Nucleotide metabolism | Purine/pyrimidine synthesis & degradation | Gout, Lesch-Nyhan syndrome, chemotherapy targets |
| Vitamins & cofactors | Fat-soluble (ADEK), water-soluble (B, C) | Scurvy, beriberi, pellagra, rickets, coagulopathy |
| Molecular biology | DNA replication, transcription, translation, repair | Xeroderma pigmentosum, Lynch syndrome, antibiotics |
02 Water, pH & Buffer Systems
Water constitutes approximately 60% of body mass and serves as the universal solvent for biochemical reactions. Its unique properties—high heat capacity, high heat of vaporization, and ability to form hydrogen bonds—make it indispensable for life. The Henderson-Hasselbalch equation (pH = pKa + log [A−]/[HA]) is the central relationship for understanding buffer chemistry and acid-base physiology.
Physiological Buffer Systems
| Buffer System | Location | pKa | Clinical Significance |
|---|---|---|---|
| Bicarbonate / CO2 | Blood (extracellular) | 6.1 | Primary extracellular buffer; regulated by lungs (CO2) and kidneys (HCO3−) |
| Phosphate (H2PO4−/HPO42−) | Intracellular, urine | 6.8 | Major intracellular buffer; important renal titratable acid |
| Hemoglobin | Red blood cells | ~7.0 | Buffering by histidine residues; Bohr effect links pH to O2 release |
| Proteins (albumin) | Blood, intracellular | Variable | Histidine, glutamate, aspartate side chains accept/donate protons |
CO2 + H2O ↔ H2CO3 ↔ H+ + HCO3−. Carbonic anhydrase catalyzes the first reaction. Despite a pKa of 6.1 (far from blood pH 7.4), this system is the most effective blood buffer because both components are independently regulated: the lungs control CO2 and the kidneys control HCO3−, making it an open buffer system.

Amino Acid Titration & pI
Each amino acid has characteristic pKa values for its α-amino group (~9.0), α-carboxyl group (~2.0), and side chain (if ionizable). The isoelectric point (pI) is the pH at which the net charge is zero. For amino acids without ionizable side chains, pI = (pKa1 + pKa2)/2. For acidic AAs (Asp, Glu), pI = average of the two lowest pKa values; for basic AAs (Lys, Arg, His), pI = average of the two highest pKa values.
Acid-Base Disorders at a Glance
| Disorder | pH | Primary Change | Compensation | Common Causes |
|---|---|---|---|---|
| Metabolic acidosis | ↓ | ↓ HCO3− | ↓ pCO2 (hyperventilation) | DKA, lactic acidosis, renal failure, diarrhea, RTA |
| Metabolic alkalosis | ↑ | ↑ HCO3− | ↑ pCO2 (hypoventilation) | Vomiting, diuretics, Cushing, Conn syndrome |
| Respiratory acidosis | ↓ | ↑ pCO2 | ↑ HCO3− (renal retention) | COPD, hypoventilation, airway obstruction, opioids |
| Respiratory alkalosis | ↑ | ↓ pCO2 | ↓ HCO3− (renal excretion) | Anxiety/hyperventilation, high altitude, PE, early sepsis |
Anion Gap
The anion gap (AG) = Na+ − (Cl− + HCO3−); normal = 8–12 mEq/L. Elevated AG metabolic acidosis indicates accumulation of unmeasured anions. Mnemonic for causes: MUDPILES — Methanol, Uremia, DKA, Propylene glycol, Iron/Isoniazid, Lactic acidosis, Ethylene glycol, Salicylates. Normal AG (hyperchloremic) acidosis: diarrhea, RTA types I & II, acetazolamide, normal saline infusion.
03 Thermodynamics & Bioenergetics
Biochemical reactions obey the laws of thermodynamics. The Gibbs free energy change (ΔG) determines whether a reaction proceeds spontaneously. ΔG < 0 indicates an exergonic (spontaneous) reaction; ΔG > 0 indicates an endergonic (nonspontaneous) reaction. ΔG depends on ΔG°′ (standard free energy change at pH 7) and the actual concentrations of reactants/products: ΔG = ΔG°′ + RT ln Q.
High-Energy Compounds
| Compound | ΔG°′ of Hydrolysis (kJ/mol) | Function |
|---|---|---|
| Phosphoenolpyruvate (PEP) | −61.9 | Highest phosphoryl-transfer potential; glycolysis substrate |
| 1,3-Bisphosphoglycerate | −49.4 | Glycolysis intermediate; substrate-level phosphorylation |
| Creatine phosphate | −43.1 | Rapid ATP regeneration in muscle and brain |
| ATP → ADP + Pi | −30.5 | Universal energy currency |
| Glucose-6-phosphate | −13.8 | Low-energy phosphate ester; traps glucose in cells |
ATP coupling allows endergonic reactions to proceed by pairing them with ATP hydrolysis so that the net ΔG is negative. Enzymes do not alter ΔG; they lower the activation energy (Ea), increasing reaction rate without shifting equilibrium.
Oxidation-Reduction Reactions
Biological oxidation-reduction (redox) reactions involve electron transfer mediated by coenzymes. NAD+/NADH and FAD/FADH2 are the major electron carriers feeding the ETC. NAD+ accepts two electrons plus a proton (as a hydride ion) to become NADH. FAD accepts two hydrogen atoms to become FADH2. The standard reduction potential (E°′) determines the direction of electron flow: electrons flow from carriers with more negative E°′ (NADH, −0.32 V) toward those with more positive E°′ (O2, +0.82 V), releasing energy captured as the proton gradient.
Shuttle Systems for Cytoplasmic NADH
The inner mitochondrial membrane is impermeable to NADH. Two shuttle systems transfer reducing equivalents from cytoplasmic NADH into the mitochondria. The malate-aspartate shuttle (liver, heart, kidney) regenerates mitochondrial NADH → 2.5 ATP/NADH. The glycerol-3-phosphate shuttle (brain, skeletal muscle) generates mitochondrial FADH2 → 1.5 ATP/NADH. This difference explains why total ATP yield from glucose is quoted as a range (30–32).
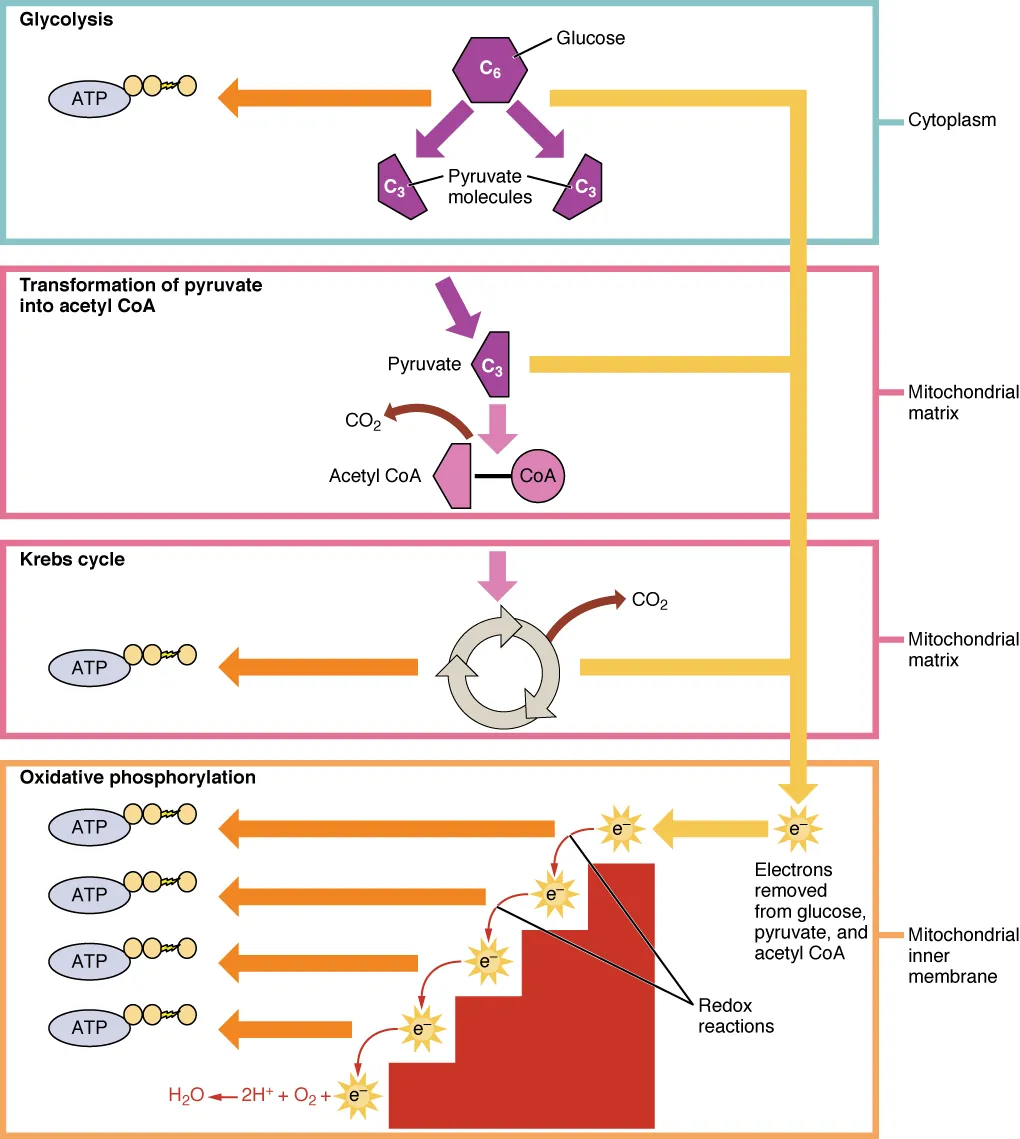
Complete oxidation of one glucose molecule: Glycolysis (2 net ATP + 2 NADH) → Pyruvate dehydrogenase (2 NADH) → TCA cycle (2 GTP + 6 NADH + 2 FADH2) → Oxidative phosphorylation yields approximately 30–32 ATP total (using the malate-aspartate shuttle for cytoplasmic NADH yields 2.5 ATP/NADH; the glycerol-3-phosphate shuttle yields 1.5 ATP/NADH).
04 Amino Acid Classification & Properties
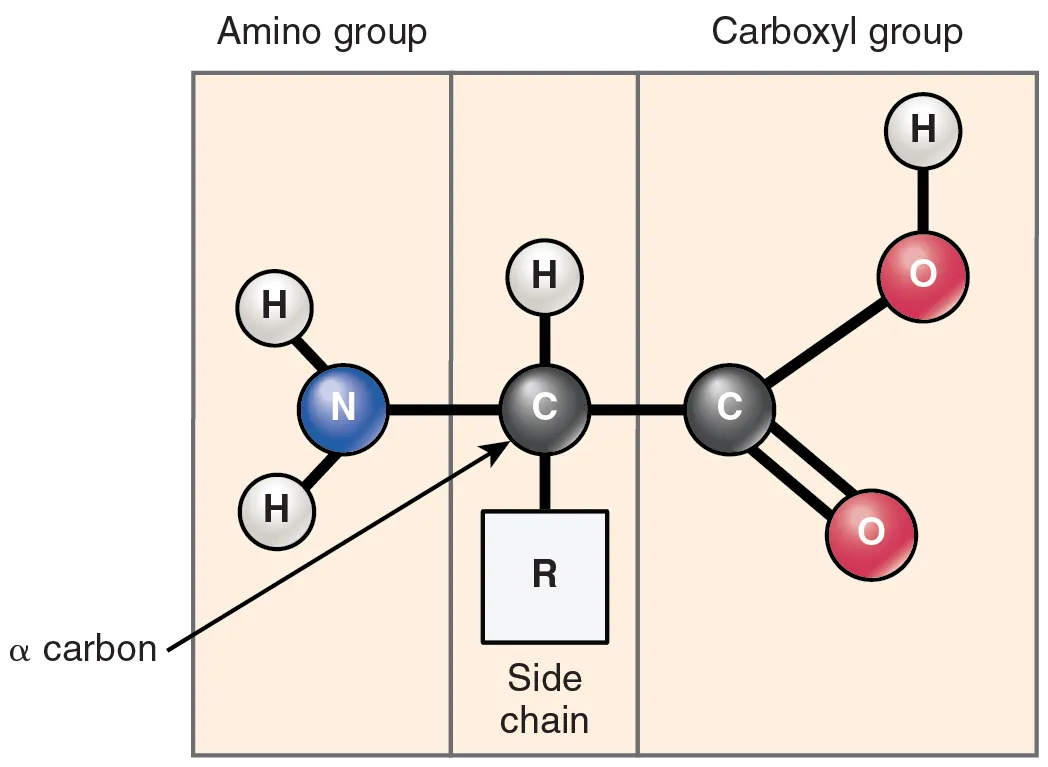
There are 20 standard amino acids encoded by the genetic code, each with a central α-carbon bonded to an amino group, a carboxyl group, a hydrogen atom, and a variable R-group (side chain). All standard amino acids except glycine are L-enantiomers. Amino acids are classified by the chemical properties of their side chains.
Essential Amino Acids
Nine amino acids cannot be synthesized by the human body and must be obtained from the diet: Phe, Val, Thr, Trp, Ile, Met, His, Leu, Lys (mnemonic: PVT TIM HaLL). Histidine is considered conditionally essential (essential in infants, marginally so in adults). Arginine is conditionally essential during growth, pregnancy, and critical illness.
Side Chain Classification
| Category | Amino Acids | Key Properties |
|---|---|---|
| Nonpolar, aliphatic | Gly, Ala, Val, Leu, Ile, Pro | Hydrophobic; found in protein interior; Pro introduces kinks (imino acid) |
| Aromatic | Phe, Tyr, Trp | UV absorption at 280 nm (Trp strongest); Tyr & Trp fluorescent |
| Polar, uncharged | Ser, Thr, Asn, Gln, Cys | H-bonding; Ser/Thr sites for phosphorylation; Cys forms disulfide bonds |
| Positively charged (basic) | Lys, Arg, His | Lys & Arg positive at pH 7.4; His partially protonated (buffer) |
| Negatively charged (acidic) | Asp, Glu | Negative at pH 7.4; transamination reactions; neurotransmitter (Glu) |
| Sulfur-containing | Cys, Met | Cys: disulfide bonds; Met: methyl donor as SAM, start codon (AUG) |
Special Amino Acids
Glycine: smallest AA, achiral, flexible; component of collagen (every 3rd residue), porphyrin synthesis, conjugation reactions. Proline: imino acid with cyclic side chain, disrupts α-helices, introduces rigid kinks; hydroxylated in collagen (requires vitamin C). Selenocysteine: the "21st amino acid," encoded by UGA with a SECIS element; found in glutathione peroxidase and thioredoxin reductase.
Amino Acid Derivatives of Clinical Importance
| Amino Acid | Derivative(s) | Clinical Relevance |
|---|---|---|
| Tryptophan | Serotonin (5-HT), melatonin, niacin (B3), NAD+ | Carcinoid syndrome: ↑ serotonin → flushing, diarrhea; tryptophan shunted away from niacin → pellagra |
| Tyrosine | Dopamine, norepinephrine, epinephrine, thyroid hormones (T3/T4), melanin | Albinism: defective tyrosinase → no melanin; Pheochromocytoma: excess catecholamine production |
| Histidine | Histamine | Allergic reactions, gastric acid secretion (H2 receptor) |
| Glutamate | GABA (via glutamic acid decarboxylase, requires B6) | Seizures in B6 deficiency (decreased GABA synthesis) |
| Glycine | Porphyrins (with succinyl-CoA), glutathione, creatine, bile acid conjugates, purines | Heme synthesis pathway; conjugation of bilirubin |
| Arginine | Nitric oxide (via NOS), creatine, urea | Endothelial vasodilation; urea cycle intermediate |
| Methionine | S-adenosylmethionine (SAM) — universal methyl donor | SAM methylates DNA, RNA, proteins, phospholipids, neurotransmitters |
05 Protein Structure & Folding
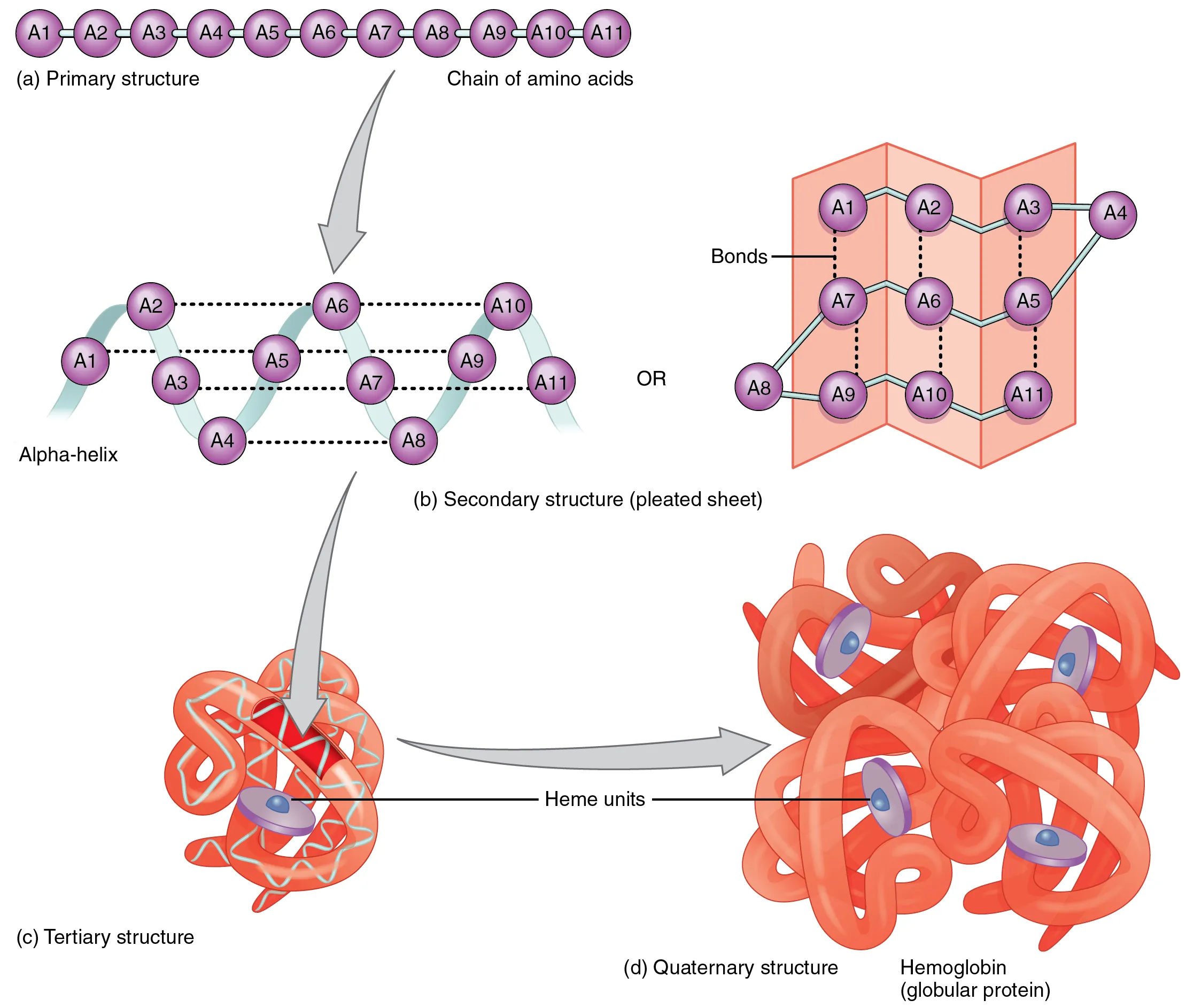
Protein function depends entirely on three-dimensional structure. There are four hierarchical levels of protein structure, each stabilized by distinct forces.
Levels of Protein Structure
| Level | Description | Stabilizing Forces | Clinical Example |
|---|---|---|---|
| Primary (1°) | Linear amino acid sequence | Peptide bonds (covalent) | Sickle cell disease: Glu6Val in β-globin |
| Secondary (2°) | α-helices, β-sheets, turns, loops | Hydrogen bonds (backbone N-H···O=C) | α-keratin (hair), β-sheet amyloid (Alzheimer) |
| Tertiary (3°) | Overall 3D shape of single polypeptide | Hydrophobic interactions, H-bonds, ionic bonds, disulfide bonds, van der Waals | Enzyme active sites; protein misfolding → prion disease |
| Quaternary (4°) | Multi-subunit assembly | Same noncovalent forces as tertiary | Hemoglobin (α2β2); antibodies |
Chaperones & Protein Folding
Chaperones assist in proper protein folding without becoming part of the final structure. Hsp70 binds hydrophobic regions of nascent polypeptides, preventing aggregation. Hsp60 (chaperonins) provide an isolated chamber for folding. Protein disulfide isomerase (PDI) catalyzes correct disulfide bond formation in the ER. Misfolded proteins are tagged with ubiquitin and degraded by the proteasome (ubiquitin-proteasome pathway).
Prion diseases (CJD, BSE): PrPC (α-helix) misfolds to PrPSc (β-sheet), which is protease-resistant and self-propagating. Amyloidosis: misfolded proteins aggregate into insoluble β-pleated sheet fibrils; stain with Congo red (apple-green birefringence under polarized light). Cystic fibrosis: ΔF508 CFTR misfolding leads to ER retention and degradation.
Post-Translational Modifications
| Modification | Residue(s) | Function |
|---|---|---|
| Phosphorylation | Ser, Thr, Tyr | Signal transduction (kinases add, phosphatases remove) |
| Glycosylation (N-linked) | Asn (in Asn-X-Ser/Thr) | Begins in ER; protein folding, stability, targeting |
| Glycosylation (O-linked) | Ser, Thr | Occurs in Golgi; mucins, blood group antigens |
| Ubiquitination | Lys | Tags proteins for proteasomal degradation |
| Methylation | Lys, Arg (histones) | Epigenetic regulation of gene expression |
| Acetylation | Lys (histones) | Histone acetylation opens chromatin (euchromatin) |
| Hydroxylation | Pro, Lys | Collagen stabilization (requires vitamin C) |
| Carboxylation | Glu | γ-carboxylation of clotting factors (requires vitamin K) |
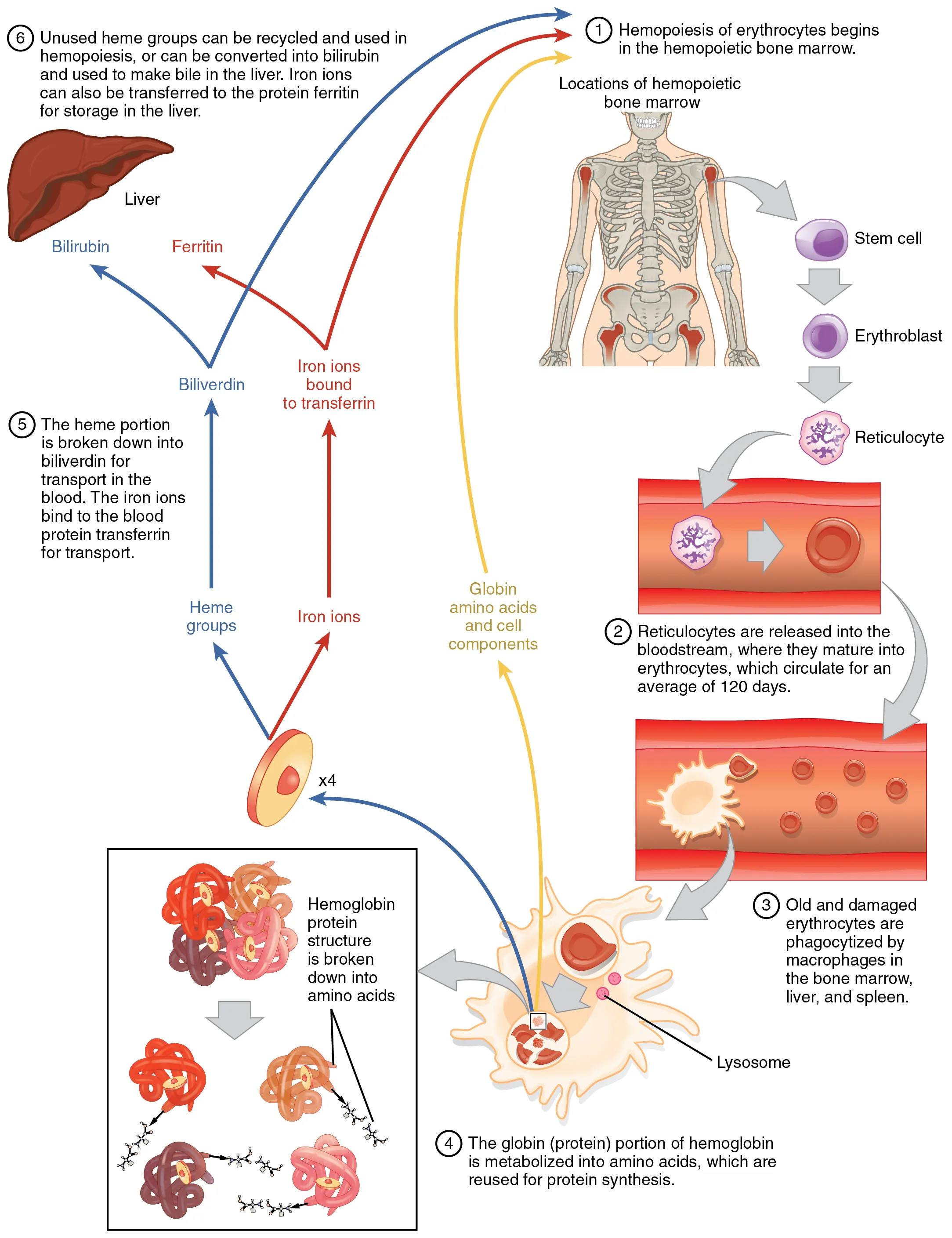
06 Hemoglobin & Oxygen Transport
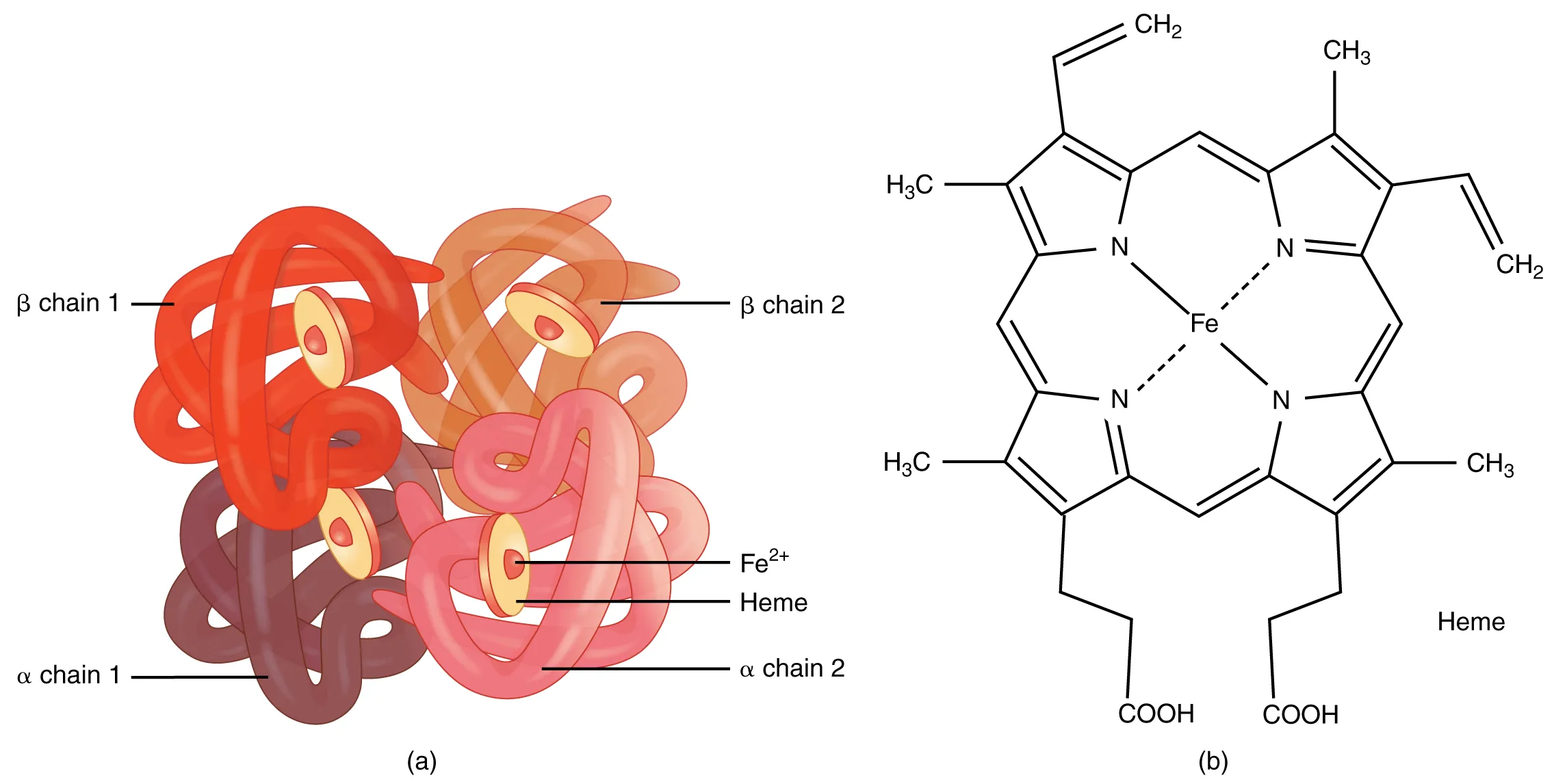
Hemoglobin (Hb) is a tetrameric protein (α2β2 in HbA) that binds up to four O2 molecules with cooperative binding, producing a sigmoidal oxygen-dissociation curve. This cooperativity arises from conformational changes between the tense (T, deoxy, low O2 affinity) and relaxed (R, oxy, high O2 affinity) states. By contrast, myoglobin is a monomer with a hyperbolic binding curve and higher O2 affinity, serving as an intracellular O2 reservoir in muscle.
Oxygen-Dissociation Curve Shifts
| Factor | Right Shift (decreased affinity → more O2 unloading) | Left Shift (increased affinity → less O2 unloading) |
|---|---|---|
| pH | ↓ pH (acidosis) — Bohr effect | ↑ pH (alkalosis) |
| CO2 | ↑ CO2 | ↓ CO2 |
| Temperature | ↑ Temperature | ↓ Temperature |
| 2,3-BPG | ↑ 2,3-BPG (altitude, anemia) | ↓ 2,3-BPG (stored blood) |
| CO / MetHb | — | CO binding & MetHb shift curve left |
| Fetal Hb | — | HbF has lower 2,3-BPG affinity → left shift |
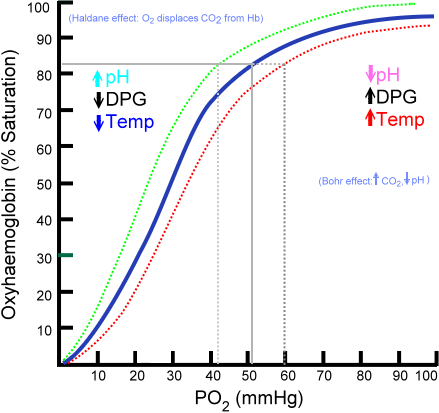
Hemoglobin Variants
HbA (α2β2): 97% of adult Hb. HbA2 (α2δ2): 2–3%; elevated in β-thalassemia trait. HbF (α2γ2): predominant in fetal life; higher O2 affinity; elevated in sickle cell treated with hydroxyurea. HbS: Glu6Val in β-globin → polymerization when deoxygenated → sickle cells. HbC: Glu6Lys → target cells. HbH (β4): 3-gene deletion α-thalassemia.
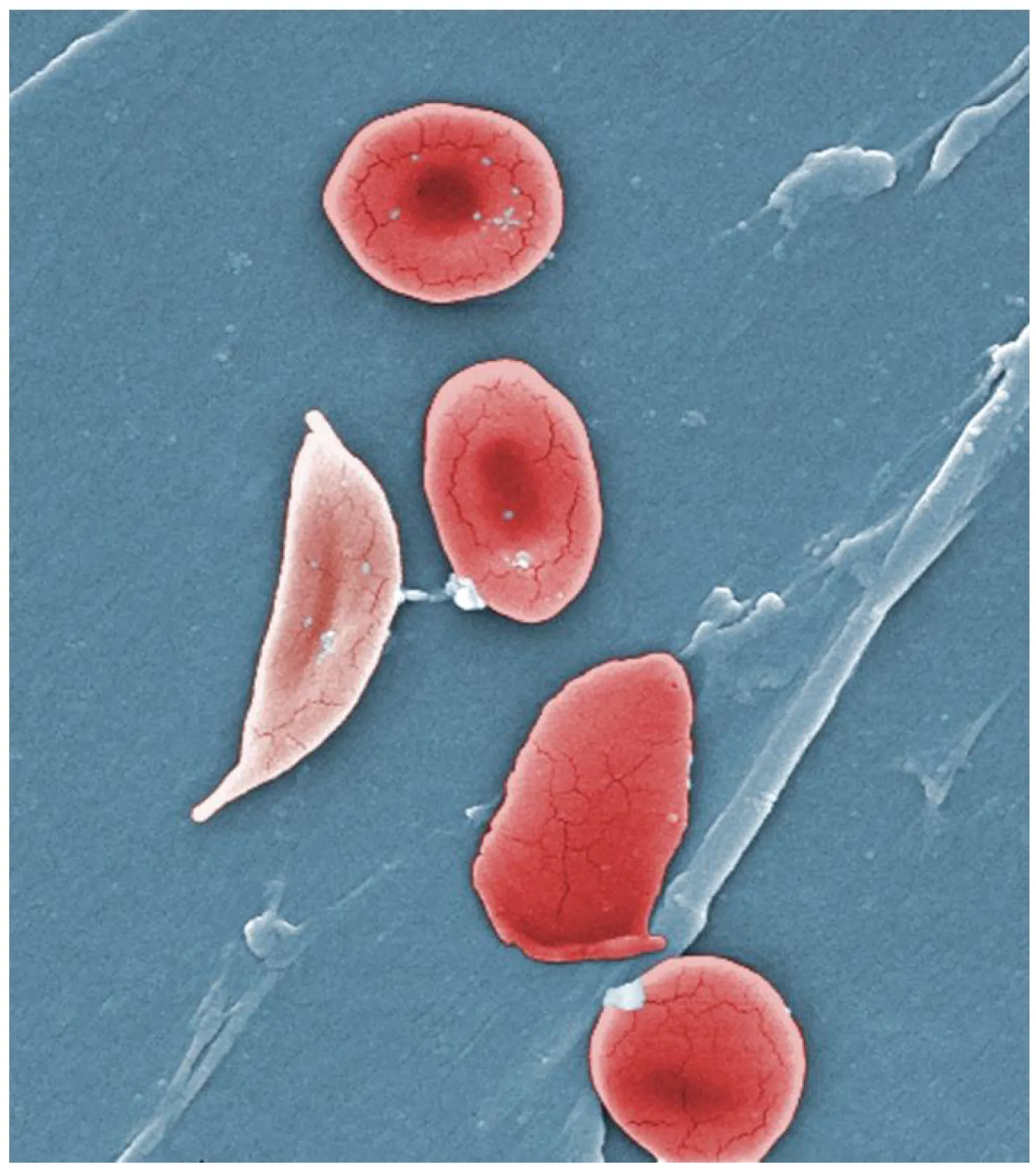
Hemoglobin Pathology Summary
| Condition | Mutation / Defect | Key Features |
|---|---|---|
| Sickle cell disease (HbSS) | Glu6Val in β-globin (AR) | Vaso-occlusive crises, acute chest syndrome, splenic sequestration/autosplenectomy, dactylitis, stroke; Tx: hydroxyurea (↑HbF), transfusions, L-glutamine, voxelotor |
| Sickle cell trait (HbAS) | One copy Glu6Val | Generally asymptomatic; protective against P. falciparum malaria; renal medullary infarcts possible at high altitude |
| α-Thalassemia | Gene deletions (chr 16), 1–4 genes | 1 gene: silent carrier. 2 genes: trait (microcytic anemia). 3 genes: HbH disease (β4 tetramers). 4 genes: hydrops fetalis (Hb Bart = γ4), fatal in utero |
| β-Thalassemia minor | One defective β-globin gene | Mild microcytic anemia, ↑HbA2 (>3.5%), target cells |
| β-Thalassemia major (Cooley) | Both β-globin genes defective | Severe anemia by 6 months, "crew-cut" skull on X-ray (extramedullary hematopoiesis), hepatosplenomegaly, iron overload from transfusions → secondary hemochromatosis |
| Methemoglobinemia | Oxidized Fe3+ in heme (cannot bind O2) | Chocolate-brown blood, cyanosis unresponsive to O2; causes: dapsone, nitrites, benzocaine; Tx: methylene blue (requires G6PD for NADPH) |
07 Collagen & Connective Tissue Disorders
Collagen is the most abundant protein in the body (~25–35% of total protein). Its hallmark is the triple helix: three left-handed polypeptide chains (each a polyproline II helix) wind around each other in a right-handed super-helix. The repeating sequence is Gly-X-Y, where X is often proline and Y is often hydroxyproline.
Collagen Types
| Type | Location | Mnemonic | Associated Disease |
|---|---|---|---|
| I | Bone, skin, tendon, dentin, cornea | "Bone" = type I | Osteogenesis imperfecta (most forms) |
| II | Cartilage, vitreous humor | "carTIIlage" | Achondrogenesis type II |
| III | Skin, blood vessels, uterus, GI (reticulin) | "three = blood vessels" | Ehlers-Danlos (vascular type IV) |
| IV | Basement membrane | "4 = floor (BM)" | Alport syndrome, Goodpasture syndrome |
Collagen Synthesis Steps
Collagen synthesis is a multi-step process spanning multiple cellular compartments:
| Step | Location | Process | Requirements / Notes |
|---|---|---|---|
| 1 | Nucleus / RER | Transcription & translation of preprocollagen | Signal peptide directs to ER |
| 2 | ER lumen | Hydroxylation of Pro & Lys residues | Vitamin C, Fe2+, α-ketoglutarate, O2 |
| 3 | ER lumen | Glycosylation of hydroxylysine residues | Specific galactosyl- and glucosyl-transferases |
| 4 | ER lumen | Triple helix formation (procollagen); disulfide bonds at C-propeptide initiate folding | Requires correct hydroxylation for stability; defective in scurvy |
| 5 | Extracellular | Cleavage of N- and C-terminal propeptides by procollagen peptidases → tropocollagen | Defective cleavage in Ehlers-Danlos type VII (dermatosparaxis) |
| 6 | Extracellular | Self-assembly into fibrils; covalent cross-linking by lysyl oxidase | Requires Cu2+; defective in Menkes disease (Cu deficiency) and lathyrism (β-aminopropionitrile inhibits lysyl oxidase) |
Osteogenesis imperfecta: defective type I collagen → brittle bones, blue sclerae, hearing loss. Ehlers-Danlos syndrome: various collagen/processing defects → hyperextensible skin, joint hypermobility; vascular type (type III collagen) → risk of arterial/organ rupture. Scurvy: vitamin C deficiency → defective hydroxylation → weak collagen → bleeding gums, petechiae, poor wound healing. Menkes disease: X-linked copper transport defect → reduced lysyl oxidase activity → connective tissue laxity, kinky hair.
Elastin & Marfan Syndrome
Elastin is an extracellular matrix protein that provides elastic recoil to tissues such as large arteries, lungs, and ligaments. It is rich in glycine, proline, and hydrophobic amino acids but (unlike collagen) is not glycosylated and has minimal hydroxyproline. Elastin monomers (tropoelastin) are cross-linked by lysyl oxidase (same enzyme as collagen cross-linking, requiring Cu2+) to form desmosine and isodesmosine cross-links unique to elastin. Marfan syndrome (AD, fibrillin-1 gene FBN1 on chromosome 15) affects the microfibrillar scaffold for elastin: tall stature, arachnodactyly, pectus deformities, upward lens subluxation, mitral valve prolapse, aortic root dilation (risk of dissection). α-1-Antitrypsin deficiency leads to unopposed elastase activity in the lungs → panacinar emphysema (lower lobes); associated with liver disease (PAS-positive, diastase-resistant globules in hepatocytes).
08 Enzyme Kinetics & Inhibition
Enzymes are biological catalysts that increase reaction rates by lowering activation energy without being consumed. Most are proteins; some are RNA (ribozymes). The active site binds substrate with specificity described by the lock-and-key model (Fischer) or the induced-fit model (Koshland). Enzyme kinetics follows the Michaelis-Menten equation: V = Vmax[S] / (Km + [S]).
Key Kinetic Parameters
| Parameter | Definition | Clinical Significance |
|---|---|---|
| Km | Substrate concentration at ½ Vmax | Reflects enzyme-substrate affinity (low Km = high affinity) |
| Vmax | Maximum reaction velocity | Proportional to enzyme concentration [ET] |
| kcat | Turnover number (Vmax/[ET]) | Catalytic efficiency of a single enzyme molecule |
| kcat/Km | Catalytic efficiency (specificity constant) | Best measure of overall enzyme efficiency |
Types of Enzyme Inhibition
| Type | Binds To | Effect on Km | Effect on Vmax | Lineweaver-Burk | Example |
|---|---|---|---|---|---|
| Competitive | Active site (competes with S) | ↑ (apparent) | Unchanged | Lines intersect on y-axis | Methotrexate vs. folate (DHFR) |
| Uncompetitive | ES complex only | ↓ (apparent) | ↓ | Parallel lines | Lithium on IMPase |
| Noncompetitive | Allosteric site (E or ES) | Unchanged | ↓ | Lines intersect on x-axis | Heavy metals on various enzymes |
| Mixed | Allosteric site (E or ES, different affinity) | Changed (up or down) | ↓ | Lines intersect in quadrant II or III | Many drug-enzyme interactions |
The double-reciprocal plot (1/V vs. 1/[S]) linearizes Michaelis-Menten kinetics. Y-intercept = 1/Vmax; X-intercept = −1/Km; Slope = Km/Vmax. Competitive inhibitors increase the x-intercept magnitude (higher apparent Km) while preserving the y-intercept (same Vmax). Overcoming competitive inhibition requires increasing [S].
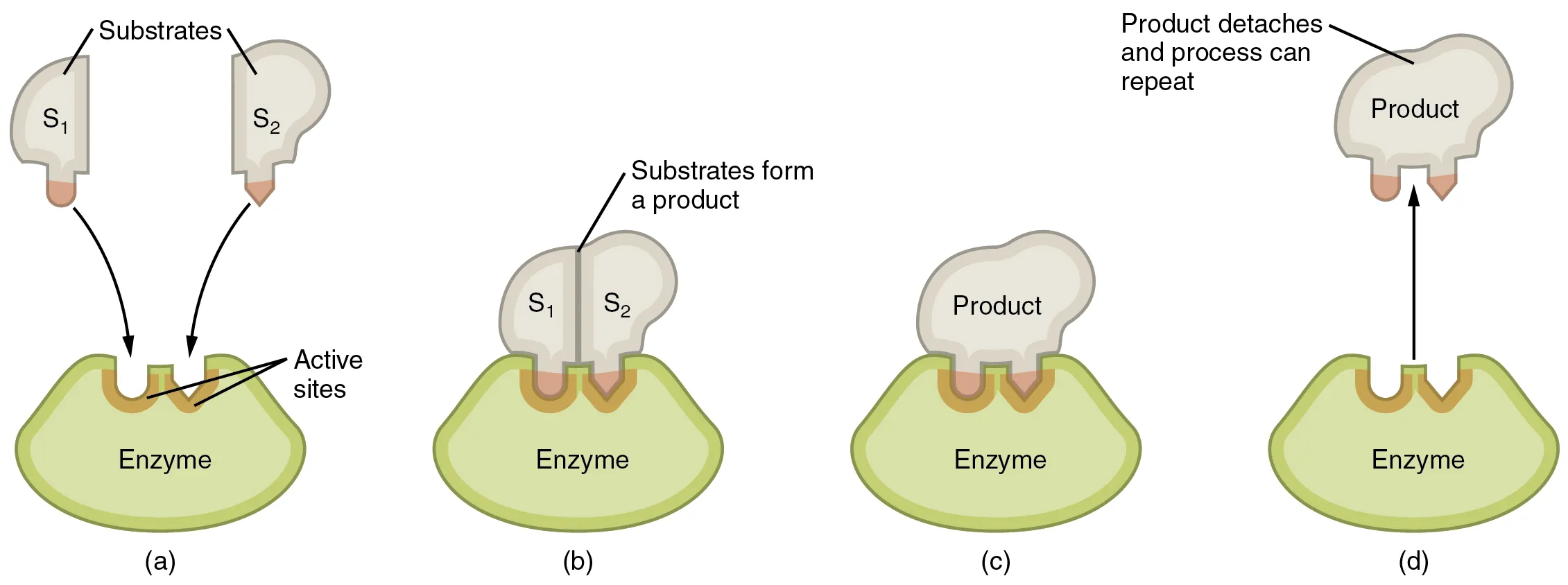
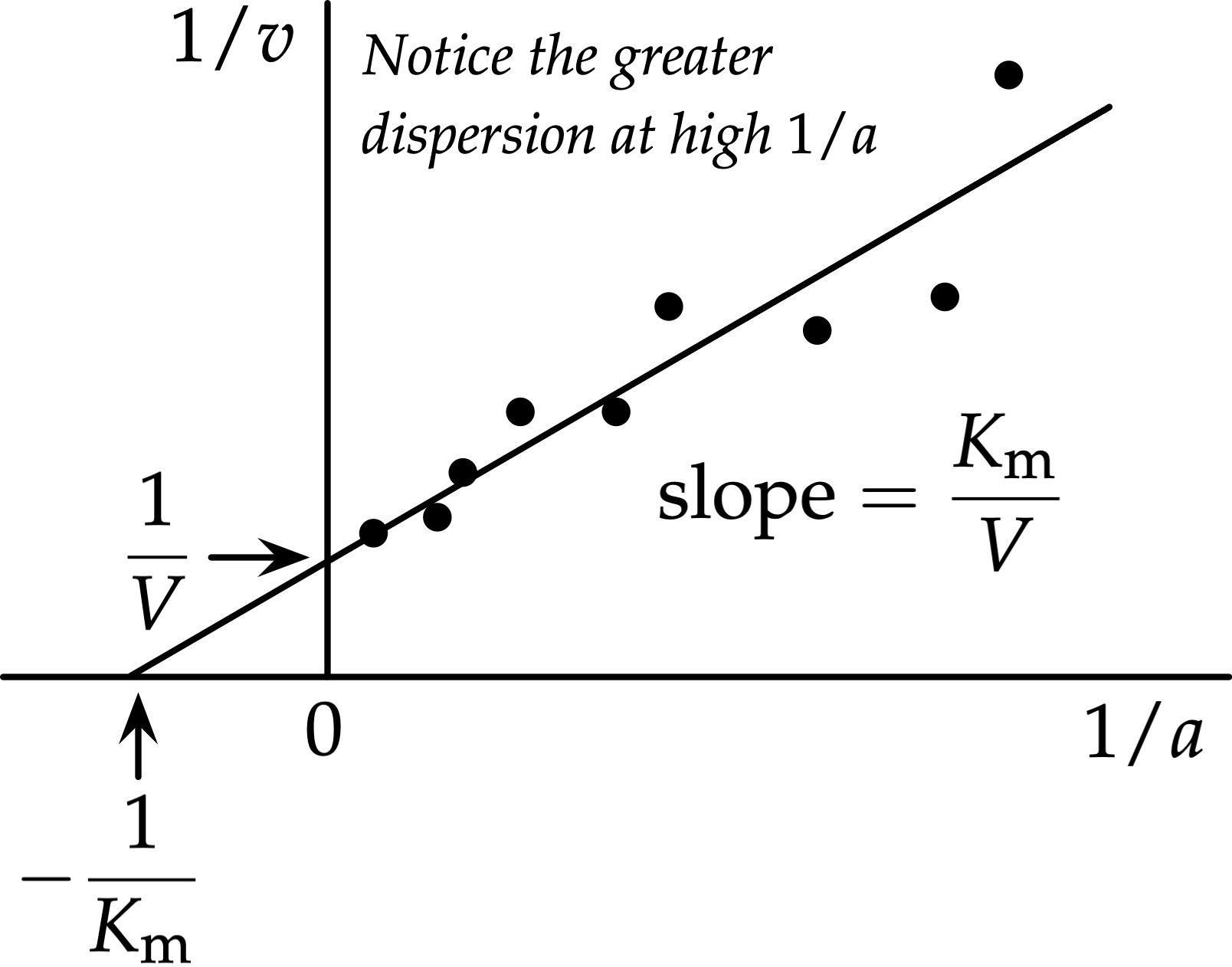
Cooperative Kinetics & Allosteric Enzymes
Allosteric enzymes do not follow Michaelis-Menten kinetics. They display a sigmoidal (S-shaped) velocity curve rather than a hyperbolic one, reflecting cooperative substrate binding among multiple subunits. The Hill coefficient (nH) quantifies cooperativity: nH > 1 = positive cooperativity (e.g., hemoglobin, nH ≈ 2.8); nH = 1 = no cooperativity (Michaelis-Menten); nH < 1 = negative cooperativity.
Allosteric activators stabilize the R (relaxed) state, shifting the curve left (lower K0.5, increased apparent affinity). Allosteric inhibitors stabilize the T (tense) state, shifting the curve right (higher K0.5, decreased apparent affinity). The classic example is PFK-1: ATP acts as both a substrate and an allosteric inhibitor at a separate regulatory site, while AMP and fructose-2,6-bisphosphate are potent allosteric activators.
Enzyme Classification (EC Numbers)
| Class | Reaction Catalyzed | Example |
|---|---|---|
| 1. Oxidoreductases | Electron transfer (oxidation/reduction) | Lactate dehydrogenase, cytochrome c oxidase |
| 2. Transferases | Transfer functional groups between molecules | Kinases (phosphoryl transfer), transaminases (amino group) |
| 3. Hydrolases | Hydrolysis (break bonds using water) | Lipases, proteases, phosphatases, esterases |
| 4. Lyases | Non-hydrolytic bond cleavage (or form double bonds) | Aldolase, decarboxylases, dehydratases |
| 5. Isomerases | Intramolecular rearrangement | Phosphoglucose isomerase, racemases, mutases |
| 6. Ligases (synthetases) | Bond formation coupled to ATP hydrolysis | DNA ligase, glutamine synthetase, acetyl-CoA carboxylase |
09 Enzyme Regulation & Clinical Enzymology
Enzyme activity is regulated at multiple levels to maintain metabolic homeostasis.
Mechanisms of Enzyme Regulation
| Mechanism | Description | Example |
|---|---|---|
| Allosteric regulation | Effector binding at a site other than the active site changes enzyme conformation | PFK-1: activated by AMP, fructose-2,6-BP; inhibited by ATP, citrate |
| Covalent modification | Phosphorylation, acetylation, etc. | Glycogen phosphorylase: active when phosphorylated (phosphorylase a) |
| Proteolytic cleavage | Irreversible activation of zymogens | Trypsinogen → trypsin; prothrombin → thrombin |
| Transcriptional control | Induction or repression of enzyme synthesis | Glucokinase induced by insulin; PEPCK induced by cortisol/glucagon |
| Compartmentalization | Separation of opposing pathways | Fatty acid synthesis (cytoplasm) vs. β-oxidation (mitochondria) |
Clinically Important Serum Enzymes
| Enzyme | Source Tissue | Clinical Use |
|---|---|---|
| AST (SGOT) | Heart, liver, muscle | MI, hepatitis; found in mitochondria + cytoplasm |
| ALT (SGPT) | Liver (most specific) | Hepatocellular injury; more liver-specific than AST |
| Alkaline phosphatase (ALP) | Bone, liver, placenta | Obstructive liver disease, bone disease (Paget) |
| GGT | Liver, biliary | Biliary disease, alcohol use; confirms hepatic origin of ↑ALP |
| Amylase / Lipase | Pancreas, salivary | Acute pancreatitis (lipase more specific & remains elevated longer) |
| Troponin I/T | Heart | Most specific and sensitive marker for MI |
| CK-MB | Heart | Reinfarction detection (rises & falls faster than troponin) |
| LDH | Ubiquitous | LDH-1 > LDH-2 in MI ("flipped ratio"); tissue ischemia/necrosis |
Isoenzymes & Isozymes
Isozymes (isoenzymes) are different molecular forms of the same enzyme that catalyze the same reaction but differ in kinetic properties, tissue distribution, and electrophoretic mobility. Clinically important examples:
| Enzyme | Isozymes | Clinical Significance |
|---|---|---|
| Lactate dehydrogenase (LDH) | LDH-1 (HHHH, heart), LDH-2 (HHHM), LDH-3, LDH-4, LDH-5 (MMMM, liver/muscle) | LDH-1 > LDH-2 = "flipped ratio" in MI; LDH-5 elevated in liver disease |
| Creatine kinase (CK) | CK-MM (muscle), CK-MB (heart), CK-BB (brain) | CK-MB elevation in MI (>5% of total CK); CK-MM in rhabdomyolysis |
| Alkaline phosphatase (ALP) | Bone, liver, intestinal, placental (Regan isozyme in cancer) | Isoform analysis distinguishes bone vs. liver source of elevation |
Enzyme Defects & Drug Metabolism
The cytochrome P450 family (particularly CYP3A4, CYP2D6, CYP2C19, CYP2C9) is responsible for phase I drug metabolism in the liver. Genetic polymorphisms cause variable drug metabolism: poor metabolizers (e.g., CYP2D6 poor metabolizers cannot activate codeine to morphine) and ultra-rapid metabolizers (excessive activation → toxicity). CYP inducers (rifampin, carbamazepine, phenobarbital, phenytoin, St. John's wort) increase drug clearance. CYP inhibitors (azole antifungals, macrolides, grapefruit juice, cimetidine, ritonavir) decrease drug clearance → toxicity risk.
Phase I (functionalization): cytochrome P450 enzymes introduce or expose a functional group (oxidation, reduction, hydrolysis) → often produces a more reactive metabolite. Phase II (conjugation): transferase enzymes attach a polar group (glucuronide, sulfate, acetyl, glutathione, glycine, methyl) → increases water solubility for renal/biliary excretion. Example: acetaminophen is conjugated (phase II) under normal conditions, but CYP2E1 (phase I) generates the toxic metabolite NAPQI, which is detoxified by glutathione conjugation. Overdose depletes glutathione → NAPQI causes hepatic necrosis. Treatment: N-acetylcysteine (replenishes glutathione).
10 Glycolysis & Pyruvate Metabolism
Glycolysis is the 10-step cytoplasmic pathway that converts one molecule of glucose into two molecules of pyruvate, generating 2 net ATP (by substrate-level phosphorylation) and 2 NADH. It is the central metabolic pathway used by all cells and the only source of ATP in cells lacking mitochondria (e.g., mature RBCs).
Key Regulatory Steps of Glycolysis
| Step | Enzyme | Reaction | Regulation |
|---|---|---|---|
| 1 | Hexokinase (or Glucokinase in liver) | Glucose → G6P | Hexokinase: inhibited by G6P; Glucokinase: induced by insulin, high Km, not inhibited by G6P |
| 3 | PFK-1 (rate-limiting) | F6P → F1,6BP | Activated by AMP, fructose-2,6-BP; Inhibited by ATP, citrate |
| 10 | Pyruvate kinase | PEP → Pyruvate | Activated by F1,6BP (feedforward); Inhibited by ATP, alanine; phosphorylated (inactivated) by glucagon |
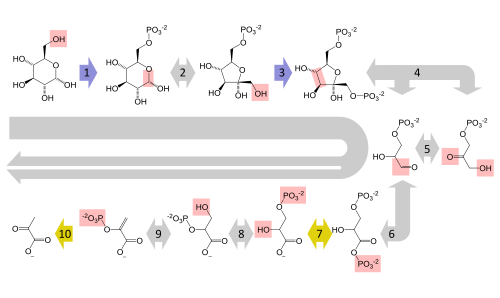
Fates of Pyruvate
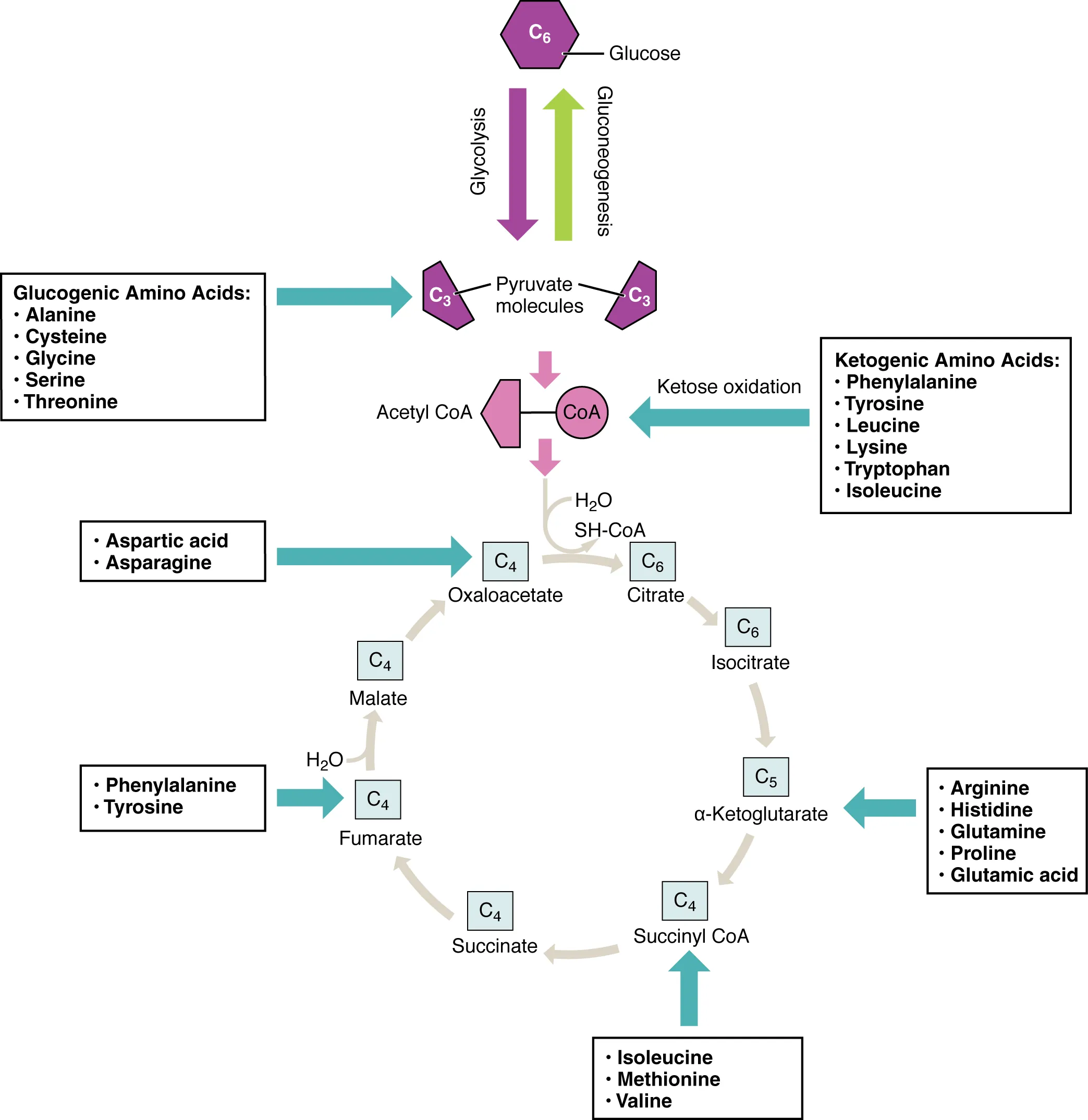
Aerobic: Pyruvate enters mitochondria → pyruvate dehydrogenase complex (PDH) converts it to acetyl-CoA + CO2 + NADH. PDH requires 5 cofactors: thiamine (B1), lipoic acid, CoA (from B5), FAD (from B2), NAD+ (from B3) (mnemonic: Tender Loving Care For Nancy). Anaerobic: Lactate dehydrogenase (LDH) converts pyruvate to lactate, regenerating NAD+ to sustain glycolysis. Transamination: Pyruvate → alanine (ALT reaction). Carboxylation: Pyruvate carboxylase converts pyruvate to oxaloacetate (gluconeogenesis entry, TCA anaplerosis).
Glucokinase vs. Hexokinase
| Feature | Hexokinase (I–III) | Glucokinase (Hexokinase IV) |
|---|---|---|
| Location | Most tissues | Liver, pancreatic β-cells |
| Km for glucose | Low (~0.1 mM) → always active at physiological glucose | High (~10 mM) → active only when glucose is elevated (fed state) |
| Vmax | Low | High (high capacity for glucose clearance) |
| Product inhibition | Inhibited by G6P | NOT inhibited by G6P |
| Induction | Constitutive | Induced by insulin |
| Clinical significance | Traps glucose in cells at all glucose levels | "Glucose sensor" in β-cells (triggers insulin secretion); MODY-2 = glucokinase mutation |
Warburg Effect
Cancer cells preferentially utilize glycolysis even in the presence of adequate O2 (aerobic glycolysis). This metabolic reprogramming supports rapid biosynthesis of nucleotides, amino acids, and lipids needed for proliferation. The Warburg effect is the basis for FDG-PET scanning in oncology: tumor cells have increased glucose uptake (via GLUT1 upregulation), and 18F-fluorodeoxyglucose accumulates because it is phosphorylated by hexokinase but cannot proceed further through glycolysis.
X-linked deficiency of E1 subunit of PDH causes lactic acidosis, neurological defects, and elevated serum alanine. Cannot convert pyruvate to acetyl-CoA, so pyruvate is shunted to lactate and alanine. Treatment: ketogenic diet (provides acetyl-CoA from fat), thiamine supplementation. Note: arsenic poisoning mimics this by inhibiting lipoic acid-containing enzymes (PDH, α-ketoglutarate dehydrogenase).
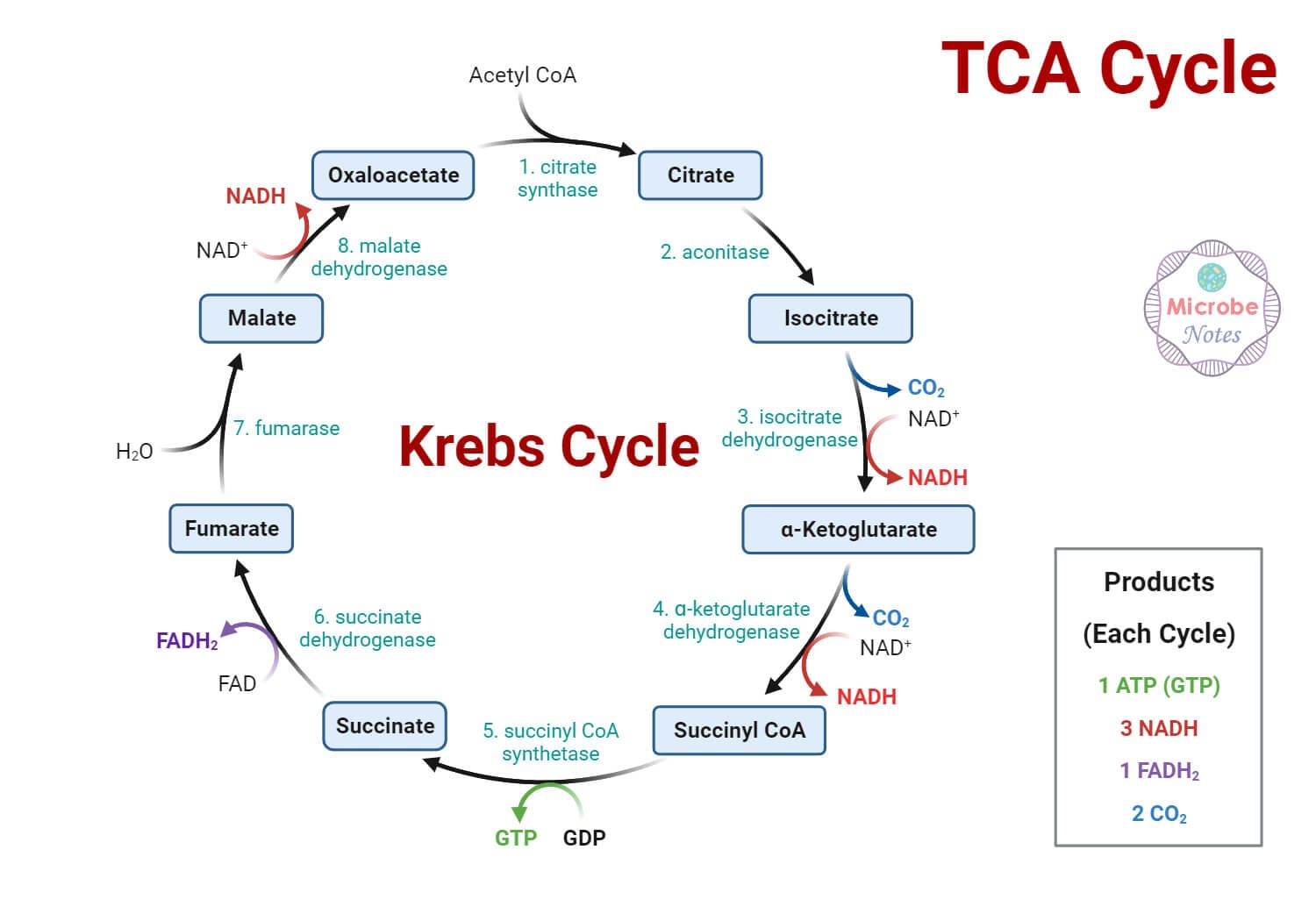
11 TCA Cycle & Oxidative Phosphorylation
The tricarboxylic acid (TCA/Krebs/citric acid) cycle occurs in the mitochondrial matrix. Acetyl-CoA condenses with oxaloacetate to form citrate, and through 8 enzymatic steps regenerates oxaloacetate while producing 3 NADH, 1 FADH2, and 1 GTP per turn. Two turns occur per glucose molecule.
TCA Cycle: Key Enzymes & Products
| Enzyme | Reaction | Product(s) | Notes |
|---|---|---|---|
| Citrate synthase | OAA + Acetyl-CoA → Citrate | Citrate | Inhibited by ATP, citrate, succinyl-CoA |
| Isocitrate dehydrogenase | Isocitrate → α-ketoglutarate | NADH, CO2 | Rate-limiting step; activated by ADP; inhibited by ATP, NADH |
| α-Ketoglutarate dehydrogenase | α-KG → Succinyl-CoA | NADH, CO2 | Same cofactors as PDH (B1, lipoate, CoA, FAD, NAD+); inhibited by succinyl-CoA, NADH |
| Succinyl-CoA synthetase | Succinyl-CoA → Succinate | GTP (substrate-level phosphorylation) | Only substrate-level phosphorylation in cycle |
| Succinate dehydrogenase | Succinate → Fumarate | FADH2 | Only enzyme embedded in inner mitochondrial membrane (Complex II of ETC) |
| Malate dehydrogenase | Malate → OAA | NADH | Regenerates OAA to continue cycle |
Electron Transport Chain & Oxidative Phosphorylation
The ETC comprises four complexes (I–IV) embedded in the inner mitochondrial membrane, plus mobile carriers (ubiquinone/CoQ, cytochrome c). NADH donates electrons at Complex I (NADH dehydrogenase); FADH2 donates at Complex II (succinate dehydrogenase). Electrons flow through Complex III (cytochrome bc1) and Complex IV (cytochrome c oxidase) to O2, reducing it to H2O. Proton pumping at Complexes I, III, and IV creates the electrochemical gradient. ATP synthase (Complex V) uses this gradient to synthesize ATP from ADP + Pi.
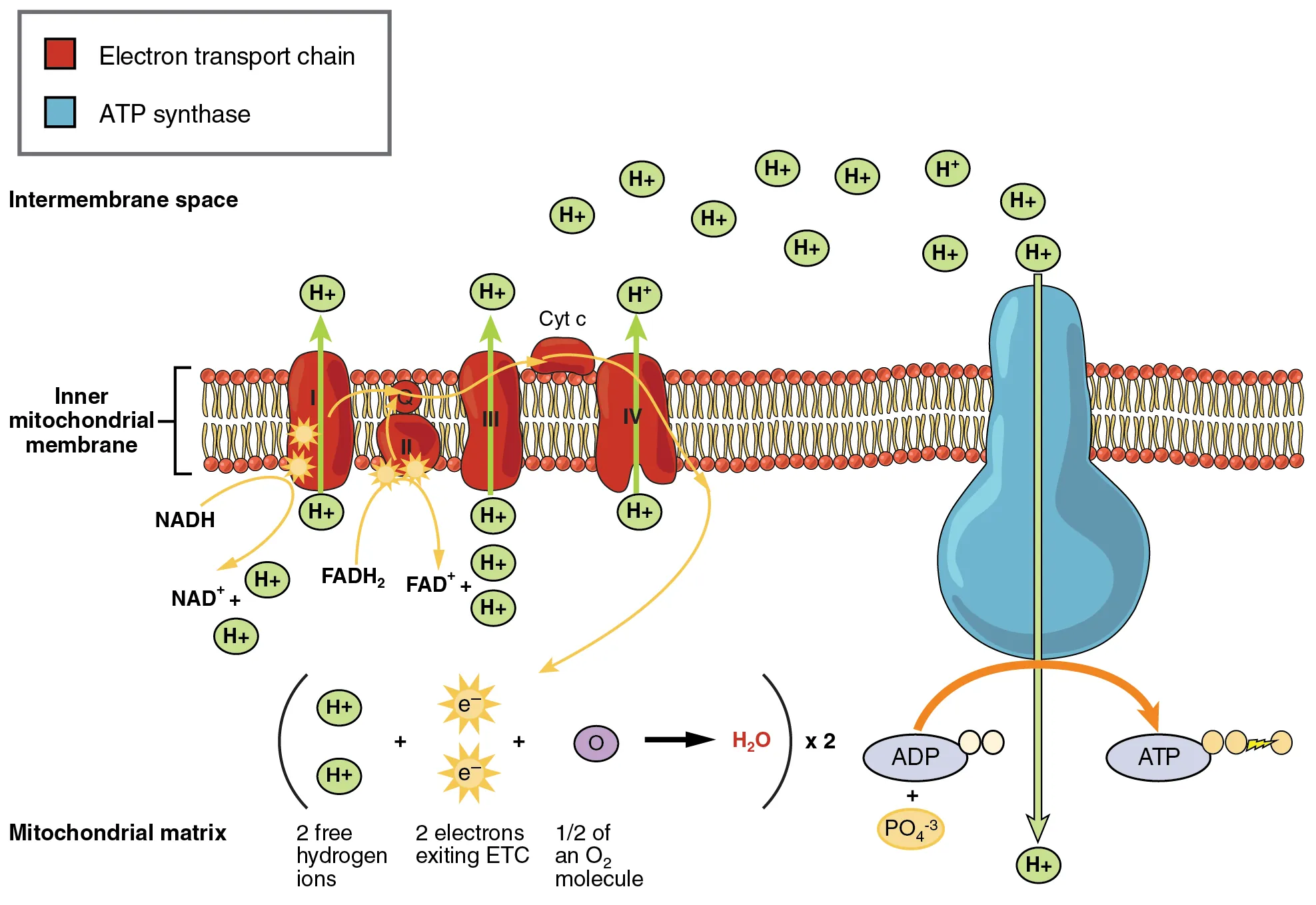
ETC Inhibitors & Uncouplers
| Agent | Target | Effect |
|---|---|---|
| Rotenone, barbiturates, piericidin A | Complex I | Block NADH → CoQ electron transfer |
| Antimycin A | Complex III | Blocks CoQ → cytochrome c transfer |
| Cyanide, CO, H2S | Complex IV | Block electron transfer to O2; lethal (histotoxic hypoxia) |
| Oligomycin | ATP synthase (Fo subunit) | Blocks proton channel; stops ATP synthesis and ETC backlog |
| 2,4-DNP, thermogenin (UCP1) | Inner membrane | Uncouple: protons leak across membrane → ↑ O2 consumption, ↓ ATP, ↑ heat |
Anaplerotic Reactions
TCA cycle intermediates are drained off for biosynthesis (e.g., citrate for fatty acid synthesis, α-KG for amino acid synthesis, succinyl-CoA for heme synthesis, OAA for gluconeogenesis). Anaplerotic reactions replenish these intermediates: the most important is pyruvate carboxylase (pyruvate + CO2 → OAA; activated by acetyl-CoA, requires biotin). Others include glutamate dehydrogenase (α-KG ↔ glutamate) and propionyl-CoA pathway (odd-chain fatty acids and some amino acids → succinyl-CoA via methylmalonyl-CoA mutase, requiring B12).
The inner mitochondrial membrane is highly selective. Key transport systems: Citrate shuttle: exports citrate to cytoplasm for fatty acid synthesis (citrate is cleaved by ATP-citrate lyase to OAA + acetyl-CoA). Malate-aspartate shuttle: transfers NADH equivalents (see bioenergetics section). Carnitine shuttle: imports long-chain fatty acids for β-oxidation. Adenine nucleotide translocase (ANT): exchanges ATP (out) for ADP (in); inhibited by atractyloside.
12 Gluconeogenesis & Glycogen Metabolism
Gluconeogenesis generates glucose from non-carbohydrate precursors (lactate, glycerol, glucogenic amino acids, propionyl-CoA) primarily in the liver and to a lesser extent the kidney cortex. It is essentially the reverse of glycolysis except at three irreversible steps, which are bypassed by different enzymes.
Gluconeogenesis Bypass Enzymes
| Glycolysis Enzyme | Gluconeogenesis Bypass | Location | Regulation |
|---|---|---|---|
| Pyruvate kinase | Pyruvate carboxylase (PYR → OAA) then PEPCK (OAA → PEP) | Mitochondria (PC) / Cytoplasm (PEPCK) | PC: activated by acetyl-CoA; PEPCK: induced by cortisol, glucagon |
| PFK-1 | Fructose-1,6-bisphosphatase | Cytoplasm | Inhibited by AMP, fructose-2,6-BP; Activated by citrate |
| Hexokinase/Glucokinase | Glucose-6-phosphatase | ER membrane (liver, kidney only) | Deficiency: von Gierke disease (GSD type I) |
Glycogen Metabolism
Glycogen Structure & Enzymes
Glycogen is a highly branched polymer of glucose with α-1,4 glycosidic bonds (linear chains) and α-1,6 glycosidic bonds (branch points, every 8–12 residues). The extensive branching creates many non-reducing ends, allowing rapid simultaneous glucose release by multiple glycogen phosphorylase molecules—critical for the rapid mobilization of glucose during exercise or the fight-or-flight response.
Glycogenesis (synthesis): Glycogenin serves as a primer (auto-glucosylating protein). Glycogen synthase (rate-limiting) adds UDP-glucose to the non-reducing end in α-1,4 linkages. Branching enzyme transfers a block of ~7 residues to create α-1,6 branch points. Glycogenolysis (breakdown): Glycogen phosphorylase (rate-limiting) cleaves α-1,4 bonds from non-reducing ends, releasing glucose-1-phosphate. Debranching enzyme has dual activity: transferase (moves 3 residues) + α-1,6-glucosidase (cleaves the branch point, releasing free glucose). In liver, G1P → G6P → free glucose (via glucose-6-phosphatase) for blood glucose maintenance; in muscle, G6P enters glycolysis directly (muscle lacks G6Pase and therefore cannot export glucose).
Insulin (fed state): activates glycogen synthase (via protein phosphatase 1), inhibits glycogen phosphorylase. Glucagon (fasting, liver): activates adenylate cyclase → cAMP → PKA → phosphorylase kinase → glycogen phosphorylase a (active). Epinephrine (muscle & liver): same cAMP-PKA cascade in liver; in muscle also via IP3 → Ca2+ → calmodulin → phosphorylase kinase activation.
Fructose-2,6-Bisphosphate: The Master Metabolic Switch
Fructose-2,6-bisphosphate (F2,6BP) is the most potent allosteric activator of PFK-1 (glycolysis) and inhibitor of fructose-1,6-bisphosphatase (gluconeogenesis). It is produced by phosphofructokinase-2 (PFK-2) and degraded by fructose-bisphosphatase-2 (FBPase-2)—both activities reside on the same bifunctional enzyme. In the fed state, insulin activates a phosphatase that dephosphorylates PFK-2/FBPase-2, promoting PFK-2 activity → ↑F2,6BP → glycolysis favored. In the fasted state, glucagon activates PKA which phosphorylates the enzyme, promoting FBPase-2 activity → ↓F2,6BP → gluconeogenesis favored.
Cori Cycle & Alanine Cycle
The Cori cycle transfers the metabolic burden of lactate recycling from muscle to liver: muscle glycolysis produces lactate → lactate travels to liver → liver converts lactate back to glucose via gluconeogenesis → glucose returns to muscle. The alanine (glucose-alanine) cycle is analogous: muscle transamination produces alanine from pyruvate → alanine travels to liver → liver converts alanine to pyruvate (then to glucose) and channels the amino group into the urea cycle. Both cycles are critical during exercise and fasting.
13 Glycogen Storage Diseases
The glycogen storage diseases (GSDs) are inherited enzyme deficiencies that lead to abnormal glycogen accumulation or impaired glycogen mobilization. They classically present with hepatomegaly, hypoglycemia, or myopathy depending on the tissue affected.
| Type | Name | Deficient Enzyme | Key Features |
|---|---|---|---|
| I | Von Gierke | Glucose-6-phosphatase | Severe fasting hypoglycemia, hepatomegaly, lactic acidosis, hyperuricemia, hyperlipidemia; liver & kidney affected |
| II | Pompe | Acid maltase (α-1,4-glucosidase, lysosomal) | Cardiomegaly (infantile form), hypotonia, macroglossia; "pomPe = Pump (heart)"; lysosomal disease |
| III | Cori (Forbes) | Debranching enzyme (α-1,6-glucosidase) | Milder form of von Gierke; short outer chains on glycogen; liver & muscle affected |
| IV | Andersen | Branching enzyme | Long, unbranched glycogen (amylopectin-like) → cirrhosis, hepatic failure; fatal in infancy without transplant |
| V | McArdle | Muscle glycogen phosphorylase | Exercise intolerance, myoglobinuria, painful cramps; "McArdle = Muscle"; no rise in blood lactate with exercise (ischemic forearm test) |
Additional Glycogen Storage Diseases
| Type | Name | Deficient Enzyme | Key Features |
|---|---|---|---|
| VI | Hers | Hepatic glycogen phosphorylase | Mild hepatomegaly, mild hypoglycemia; benign course; "Hers = Hepatic" |
| VII | Tarui | Muscle PFK-1 | Exercise intolerance similar to McArdle; hemolytic anemia (PFK-1 also in RBCs) |
| IX | — | Phosphorylase kinase | Most common GSD overall; X-linked or AR forms; hepatomegaly, mild hypoglycemia, growth delay; excellent prognosis |
GSDs presenting primarily with hepatomegaly & hypoglycemia: von Gierke (I), Cori (III), Hers (VI), phosphorylase kinase deficiency (IX). GSDs presenting primarily with myopathy: McArdle (V), Tarui (VII). Pompe (II) is unique: it is a lysosomal storage disease (acid maltase) causing cardiomyopathy and skeletal myopathy. Key lab differentiators: von Gierke has lactic acidosis, hyperuricemia, and hyperlipidemia (the others do not to the same degree).
14 Pentose Phosphate Pathway & G6PD Deficiency
The pentose phosphate pathway (PPP/HMP shunt) occurs in the cytoplasm and has two phases. The oxidative phase is irreversible and generates NADPH (for reductive biosynthesis and glutathione reduction) and ribulose-5-phosphate. The non-oxidative phase is reversible, interconverting sugars and connecting to glycolysis (via fructose-6-phosphate and glyceraldehyde-3-phosphate). The rate-limiting enzyme is glucose-6-phosphate dehydrogenase (G6PD).
Pentose Phosphate Pathway — Two Phases
The oxidative phase is irreversible and consists of three reactions: (1) G6PD oxidizes G6P to 6-phosphoglucono-δ-lactone (producing the first NADPH). (2) Lactonase hydrolyzes the lactone to 6-phosphogluconate. (3) 6-Phosphogluconate dehydrogenase oxidizes and decarboxylates to ribulose-5-phosphate (producing the second NADPH + CO2). Net per G6P entering: 2 NADPH + 1 ribulose-5-phosphate + 1 CO2.
The non-oxidative phase is reversible and uses transketolase (requires TPP/B1) and transaldolase to interconvert C3, C4, C5, C6, and C7 sugars. This connects the PPP to glycolysis: it can generate ribose-5-phosphate for nucleotide synthesis when needed, or funnel excess pentose phosphates back into glycolysis (as F6P and G3P) when NADPH demand exceeds ribose demand. Tissues with high NADPH demand (liver, adrenal cortex, RBCs, lactating mammary gland, adipose) have particularly active PPP.
Functions of NADPH
| Process | NADPH Role | Clinical Significance |
|---|---|---|
| Glutathione reduction | Glutathione reductase uses NADPH to regenerate reduced glutathione (GSH) | GSH detoxifies H2O2 in RBCs; deficiency → oxidative hemolysis |
| Fatty acid synthesis | Reductive biosynthesis | Lipogenesis in liver, adipose, lactating mammary gland |
| Cholesterol synthesis | HMG-CoA reductase uses NADPH | Target of statins |
| Steroid synthesis | Cytochrome P450 reactions | Adrenal steroidogenesis |
| Respiratory burst | NADPH oxidase generates superoxide (O2−) | Defective in chronic granulomatous disease (CGD) |
| Nitric oxide synthesis | NOS requires NADPH | Endothelial vasodilation |
G6PD Deficiency
G6PD deficiency is the most common enzyme deficiency worldwide (X-linked recessive, protective against P. falciparum malaria). Reduced NADPH production leads to inability to maintain reduced glutathione in RBCs → oxidative damage to hemoglobin → Heinz bodies (denatured Hb precipitates) and bite cells (splenic macrophage removal of Heinz bodies). Episodes are triggered by oxidant stresses: fava beans, infections, sulfonamides, primaquine, dapsone, nitrofurantoin.
G6PD Deficiency Variants
| Variant | Population | Enzyme Activity | Clinical Severity |
|---|---|---|---|
| G6PD B (wild type) | Most populations | Normal (100%) | None |
| G6PD A+ (normal variant) | African descent | 85–100% | None |
| G6PD A− | African descent (10–15%) | 5–15% | Mild/moderate episodic hemolysis; self-limited (older RBCs destroyed, reticulocytes have adequate enzyme) |
| G6PD Mediterranean | Mediterranean, Middle Eastern | <1% | Severe; hemolysis can be life-threatening; all RBC ages affected; favism (fava beans) |
Oxidant Stressors Triggering G6PD Hemolysis
Common triggers include: Infections (most common precipitant overall), fava beans (contain vicine/divicine), medications (primaquine, dapsone, sulfonamides, nitrofurantoin, rasburicase, methylene blue), and metabolic acidosis (DKA). Methylene blue is both a cause (oxidant in excess) and a treatment for methemoglobinemia (requires NADPH from G6PD to reduce MetHb), meaning methylene blue is contraindicated in G6PD-deficient patients with methemoglobinemia—use ascorbic acid instead.
15 Fructose & Galactose Metabolism
Fructose and galactose are dietary sugars that enter glycolysis after conversion to glycolytic intermediates. Defects in these pathways cause distinct clinical syndromes.
Fructose Metabolism Disorders
| Condition | Deficient Enzyme | Accumulated Substrate | Clinical Features |
|---|---|---|---|
| Essential fructosuria | Fructokinase | Fructose (in blood & urine) | Benign; asymptomatic; incidental finding |
| Hereditary fructose intolerance | Aldolase B | Fructose-1-phosphate | Hypoglycemia, jaundice, vomiting after fructose/sucrose ingestion; F1P inhibits glycogenolysis & gluconeogenesis; traps phosphate → ↓ATP |
Galactose Metabolism Disorders
| Condition | Deficient Enzyme | Accumulated Substrate | Clinical Features |
|---|---|---|---|
| Galactokinase deficiency | Galactokinase | Galactitol | Infantile cataracts (galactitol osmotic damage); relatively mild |
| Classic galactosemia | Galactose-1-phosphate uridylyltransferase | Galactose-1-phosphate, galactitol | Failure to thrive, jaundice, hepatomegaly, infantile cataracts, intellectual disability, E. coli sepsis in neonates; treatment: eliminate galactose & lactose from diet |
Both fructose and galactose disorders illustrate a common biochemical principle: when an enzyme in a pathway is deficient, its substrate accumulates and is shunted into alternative pathways (e.g., aldose reductase converts galactose to galactitol, which accumulates in the lens causing cataracts). This "upstream backup and side-shunt" pattern recurs throughout metabolic disease.
Sorbitol (Polyol) Pathway
In tissues that do not require insulin for glucose uptake (lens, retina, kidneys, Schwann cells), chronically elevated glucose is converted to sorbitol by aldose reductase (using NADPH) and then to fructose by sorbitol dehydrogenase (using NAD+). Because sorbitol cannot cross cell membranes, it accumulates intracellularly, causing osmotic damage. This mechanism contributes to diabetic complications: cataracts (lens), retinopathy (retina), peripheral neuropathy (Schwann cells), and nephropathy (kidney). Aldose reductase inhibitors have been explored therapeutically but have limited clinical success to date.
Lactose Intolerance
Lactase is a brush border enzyme in small intestinal villi that cleaves lactose into glucose + galactose. Lactase deficiency (primary: age-related downregulation, most common worldwide; secondary: mucosal injury from celiac disease, infectious enteritis, etc.) leads to undigested lactose reaching the colon → bacterial fermentation → bloating, flatulence, osmotic diarrhea, abdominal cramps. Diagnosis: hydrogen breath test (colonic bacteria metabolize lactose producing H2). Treatment: dietary lactose restriction, oral lactase supplements.
Other Disaccharidase Deficiencies
In addition to lactase, the brush border contains sucrase-isomaltase (cleaves sucrose into glucose + fructose, and isomaltose at α-1,6 branch points) and trehalase (cleaves trehalose from mushrooms). Congenital sucrase-isomaltase deficiency presents in infancy with osmotic diarrhea upon introduction of sucrose-containing foods (fruits, juice, table sugar). Secondary disaccharidase deficiency can accompany any condition damaging the intestinal brush border (viral gastroenteritis, celiac disease, Crohn disease, tropical sprue). After mucosal recovery, disaccharidase activity typically returns, but lactase is the slowest to recover and may remain persistently low.
16 Fatty Acid Synthesis & Beta-Oxidation
Fatty acid synthesis and β-oxidation are reciprocally regulated pathways that occur in different cellular compartments.
Fatty Acid Synthesis vs. Beta-Oxidation
| Feature | Synthesis | β-Oxidation |
|---|---|---|
| Location | Cytoplasm | Mitochondrial matrix |
| Carrier | ACP (acyl carrier protein) | CoA |
| Key enzyme | Acetyl-CoA carboxylase (ACC) — rate-limiting | Carnitine palmitoyltransferase I (CPT-I) — rate-limiting |
| Carbon units | Adds 2C per cycle (from malonyl-CoA) | Removes 2C per cycle (as acetyl-CoA) |
| Reducing agent | NADPH (from PPP, malic enzyme) | Produces NADH & FADH2 |
| Activated by | Insulin, citrate | Glucagon, epinephrine |
| Inhibited by | Glucagon (phosphorylation of ACC), palmitoyl-CoA | Malonyl-CoA (inhibits CPT-I) |
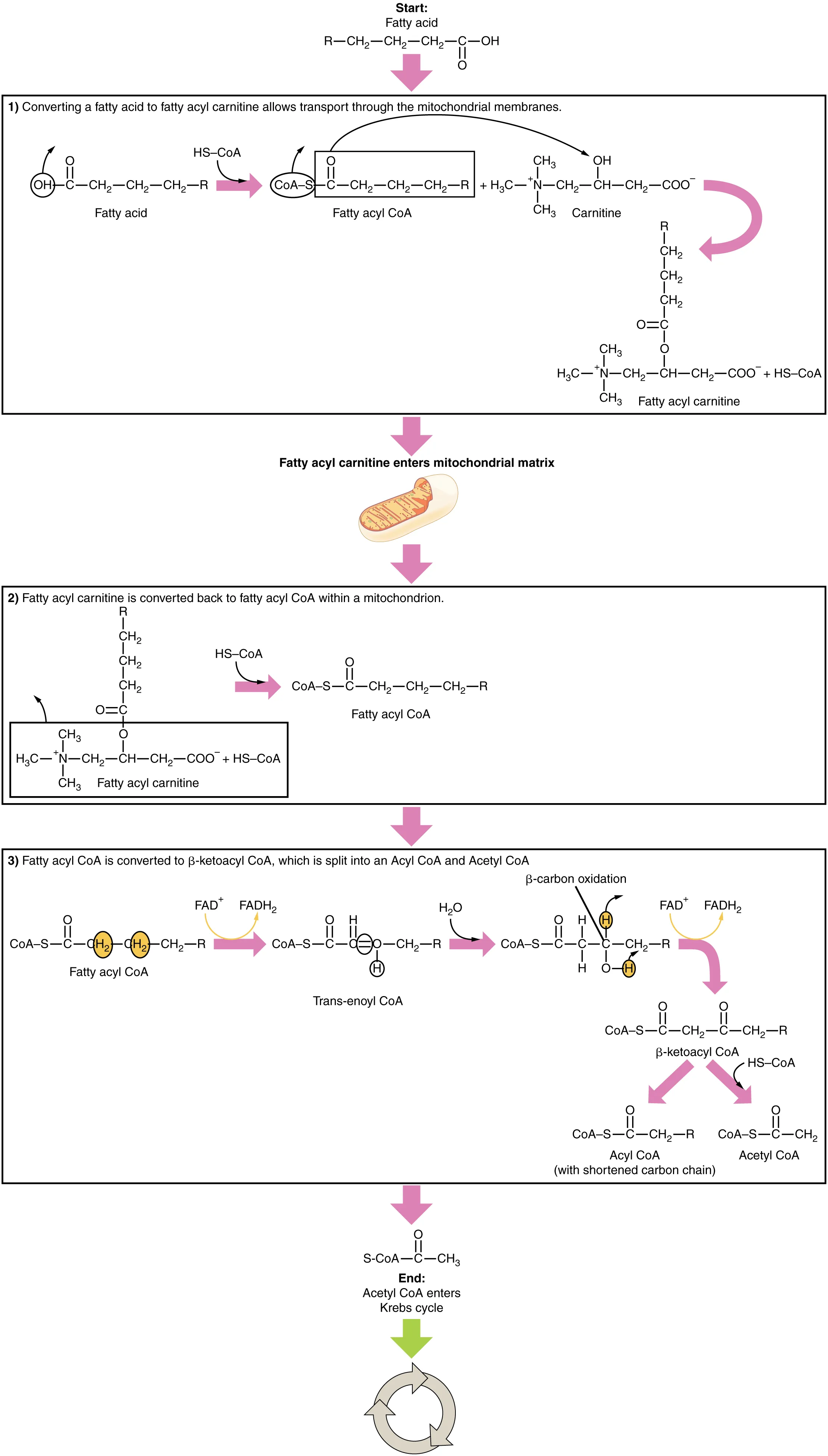
Carnitine Shuttle
Long-chain fatty acids cannot cross the inner mitochondrial membrane directly. The carnitine shuttle is a three-step process:
| Step | Enzyme / Transporter | Location | Reaction |
|---|---|---|---|
| 1 | CPT-I (carnitine palmitoyltransferase I) | Outer mitochondrial membrane | Acyl-CoA + carnitine → acylcarnitine + CoA |
| 2 | Carnitine-acylcarnitine translocase | Inner mitochondrial membrane | Acylcarnitine enters; free carnitine exits |
| 3 | CPT-II (carnitine palmitoyltransferase II) | Inner mitochondrial membrane (matrix side) | Acylcarnitine + CoA → acyl-CoA + carnitine |
CPT-I is the rate-limiting step and the key regulatory point: it is allosterically inhibited by malonyl-CoA (the first committed intermediate in fatty acid synthesis). This ensures that synthesis and oxidation do not occur simultaneously. Medium-chain fatty acids (C6–C12) bypass the carnitine shuttle entirely, entering the mitochondrial matrix by diffusion.
Fatty Acid Oxidation Disorders
Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency is the most common fatty acid oxidation defect. Presents in infancy with hypoketotic hypoglycemia, vomiting, lethargy, and liver dysfunction after fasting. Elevated medium-chain acylcarnitines on newborn screen. Avoid fasting; frequent feeding. Systemic carnitine deficiency presents similarly; treatment: oral carnitine supplementation.
Odd-Chain & Very-Long-Chain Fatty Acid Oxidation
Odd-chain fatty acids (e.g., C15, C17, found in dairy and ruminant fat) undergo β-oxidation normally until the final 3-carbon unit, propionyl-CoA. Propionyl-CoA carboxylase (requires biotin) converts it to D-methylmalonyl-CoA, which is racemized and then converted to succinyl-CoA by methylmalonyl-CoA mutase (requires B12). Succinyl-CoA enters the TCA cycle. B12 deficiency thus causes elevated methylmalonic acid.
Very-long-chain fatty acids (VLCFAs, ≥C22) are first shortened in peroxisomes (via peroxisomal β-oxidation), then transferred to mitochondria for completion. Zellweger syndrome: absent peroxisomes (peroxisome biogenesis disorder) → VLCFA accumulation, hypotonia, seizures, hepatomegaly, characteristic facies; fatal in infancy. X-linked adrenoleukodystrophy: defective peroxisomal VLCFA transporter (ABCD1) → VLCFA accumulation in adrenal cortex and CNS white matter → adrenal insufficiency, progressive demyelination.
17 Ketogenesis & Ketolysis
Ketogenesis occurs in liver mitochondria during prolonged fasting, starvation, or uncontrolled diabetes when acetyl-CoA production (from β-oxidation) exceeds TCA cycle capacity. The three ketone bodies are acetoacetate, β-hydroxybutyrate (the predominant circulating form), and acetone (volatile, exhaled → fruity breath).
Ketogenesis Pathway
2 Acetyl-CoA → acetoacetyl-CoA (thiolase) → HMG-CoA (HMG-CoA synthase, rate-limiting) → acetoacetate + acetyl-CoA (HMG-CoA lyase). Acetoacetate is reduced to β-hydroxybutyrate by β-hydroxybutyrate dehydrogenase (using NADH), or spontaneously decarboxylated to acetone.
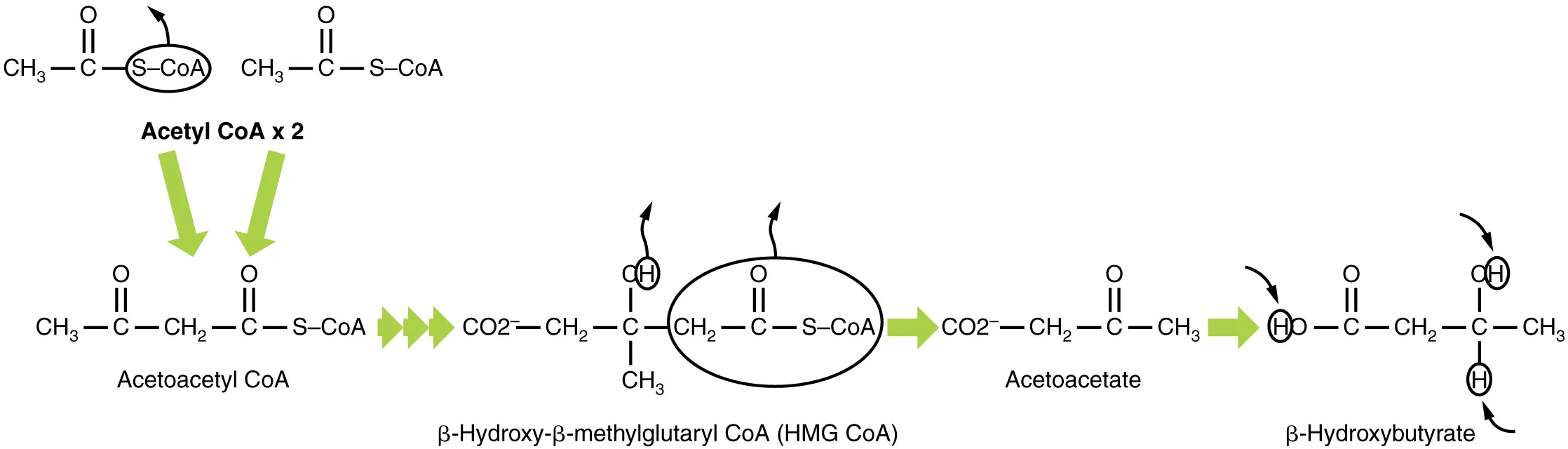
Ketolysis (Peripheral Utilization)
Extrahepatic tissues (brain, heart, skeletal muscle, kidney) convert β-hydroxybutyrate back to acetoacetate, then to acetoacetyl-CoA via succinyl-CoA-acetoacetate CoA transferase (thiophorase), and finally to 2 acetyl-CoA for the TCA cycle. The liver cannot use ketone bodies because it lacks thiophorase.
Uncontrolled type 1 diabetes → absolute insulin deficiency → unopposed lipolysis → massive β-oxidation → excess acetyl-CoA → ketone body overproduction. Accumulation of acetoacetate and β-hydroxybutyrate causes high-anion-gap metabolic acidosis. Presentation: Kussmaul breathing (compensatory hyperventilation), fruity breath (acetone), abdominal pain, dehydration. Lab: high glucose, high anion gap, low bicarbonate, positive serum ketones. Treatment: IV fluids, insulin, potassium replacement, monitoring.
Alcoholic Ketoacidosis (AKA)
Chronic alcohol use combined with poor nutrition and recent binge → depleted glycogen stores + ↑NADH/NAD+ ratio (from ethanol metabolism) → impaired gluconeogenesis + enhanced lipolysis and ketogenesis. Unlike DKA, glucose is typically low or normal (not elevated). Labs show high anion gap metabolic acidosis with positive ketones. β-Hydroxybutyrate predominates (high NADH shifts acetoacetate → β-HB), so the nitroprusside test (detects only acetoacetate) may be falsely negative initially. Treatment: IV fluids with dextrose (D5NS), thiamine before glucose (to prevent Wernicke encephalopathy), electrolyte replacement.
Starvation Ketosis
During prolonged fasting (>48–72 hours), hepatic glycogen is exhausted and gluconeogenesis alone cannot meet glucose demands. The liver increases ketogenesis from fatty acids. Ketone bodies (particularly β-hydroxybutyrate) become the brain's major fuel source, reducing the need for gluconeogenesis and thereby sparing muscle protein. In simple starvation, the degree of ketoacidosis is milder than in DKA because basal insulin secretion is preserved, limiting lipolysis to a controlled rate.
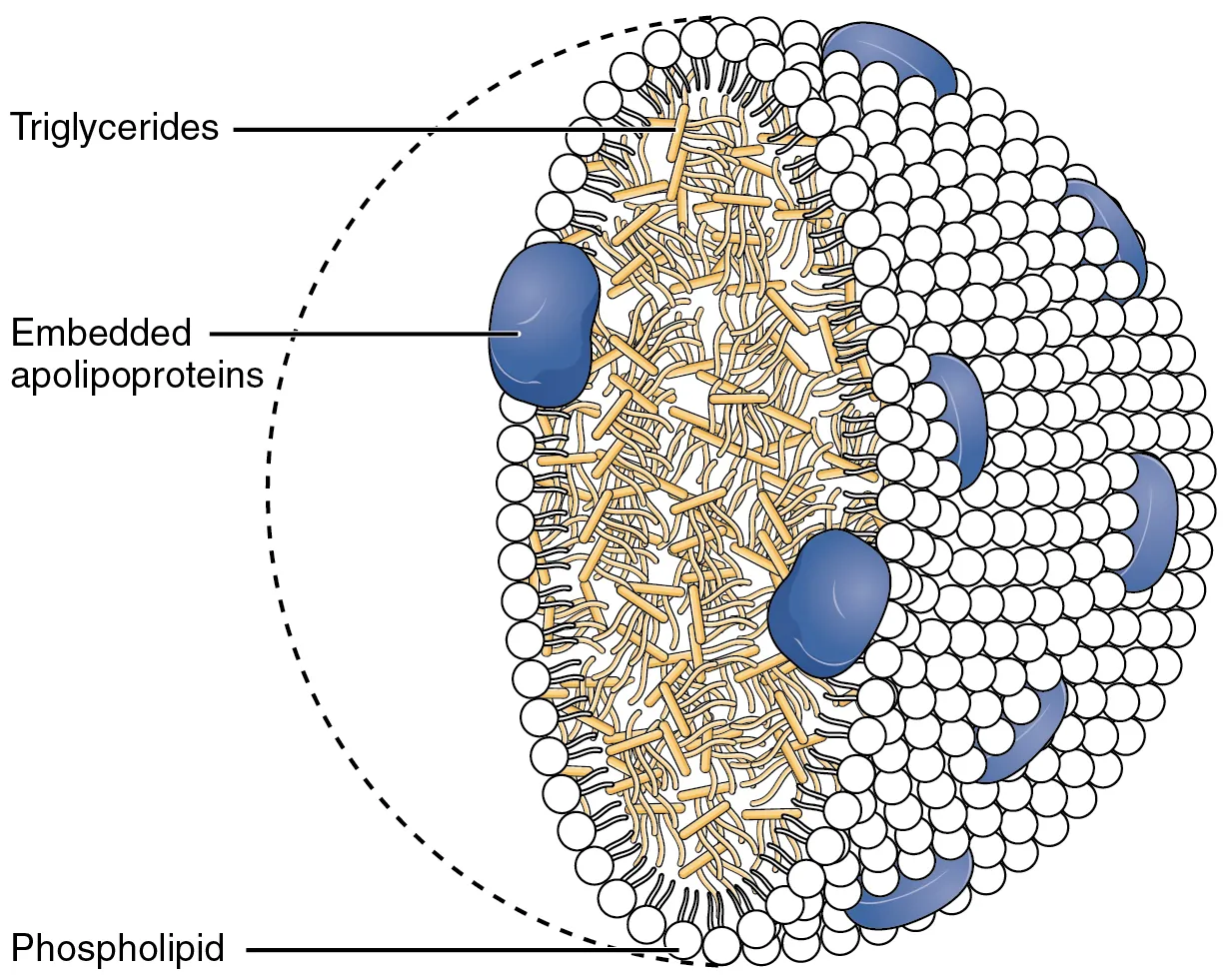
18 Lipoproteins & Cholesterol Metabolism
Lipids are transported in blood as lipoproteins—spherical particles with a hydrophobic core (triglycerides, cholesterol esters) and an amphipathic shell (phospholipids, free cholesterol, apolipoproteins).
Lipoprotein Classes
| Lipoprotein | Source | Major Lipid | Key Apolipoprotein | Function |
|---|---|---|---|---|
| Chylomicrons | Intestine | Dietary TGs | ApoB-48, ApoC-II, ApoE | Deliver dietary TGs to peripheral tissues |
| VLDL | Liver | Hepatic TGs | ApoB-100, ApoC-II, ApoE | Deliver endogenous TGs to peripheral tissues |
| IDL | VLDL remnant | TGs + cholesterol | ApoB-100, ApoE | Intermediate; converted to LDL or taken up by liver |
| LDL | IDL conversion | Cholesterol | ApoB-100 | Delivers cholesterol to tissues; "bad cholesterol" |
| HDL | Liver, intestine | Cholesterol esters | ApoA-I, ApoC-II, ApoE | Reverse cholesterol transport; "good cholesterol" |
Key Enzymes & Receptors
Lipoprotein lipase (LPL): on capillary endothelium; activated by ApoC-II; hydrolyzes TGs in chylomicrons and VLDL. Hepatic lipase: converts IDL to LDL. LCAT (lecithin-cholesterol acyltransferase): esterifies cholesterol on HDL; activated by ApoA-I. CETP (cholesterol ester transfer protein): transfers cholesterol esters from HDL to LDL/VLDL in exchange for TGs. LDL receptor: hepatic uptake of LDL; recognizes ApoB-100 and ApoE.
Dyslipidemias
| Condition | Defect | Lipid Profile | Clinical Features |
|---|---|---|---|
| Familial hypercholesterolemia (type IIa) | LDL receptor deficiency (AD) | ↑↑↑ LDL | Xanthomas (tendon), xanthelasma, corneal arcus, early MI |
| Familial hypertriglyceridemia (type I) | LPL or ApoC-II deficiency | ↑↑↑ TGs, ↑ chylomicrons | Pancreatitis, eruptive xanthomas, lipemia retinalis, creamy supernatant |
| Familial dysbetalipoproteinemia (type III) | ApoE2/E2 homozygosity | ↑ TGs, ↑ cholesterol, ↑ IDL | Palmar xanthomas, tuberoeruptive xanthomas |
| Abetalipoproteinemia | MTP deficiency (no ApoB-48/100) | ↓↓ all lipoproteins with ApoB | Fat malabsorption, acanthocytosis, retinitis pigmentosa, ataxia |
Cholesterol Synthesis & Bile Acid Metabolism
Cholesterol is synthesized from acetyl-CoA in the liver. The rate-limiting enzyme is HMG-CoA reductase (converts HMG-CoA to mevalonate). This enzyme is the target of statins. Cholesterol is the precursor for bile acids (7α-hydroxylase is rate-limiting), steroid hormones (all classes), and vitamin D. Bile acids (cholic, chenodeoxycholic) are conjugated with glycine or taurine, secreted into bile, and recycled via the enterohepatic circulation (terminal ileum reabsorption). Bile acid sequestrants (cholestyramine) interrupt this cycle and are used to lower LDL cholesterol and treat bile acid diarrhea.
Eicosanoid Synthesis
Eicosanoids (prostaglandins, thromboxanes, leukotrienes) are derived from arachidonic acid (a 20-carbon polyunsaturated fatty acid released from membrane phospholipids by phospholipase A2). COX-1/COX-2 produce prostaglandins and thromboxanes (NSAIDs and aspirin inhibit COX). 5-Lipoxygenase produces leukotrienes (zileuton inhibits; montelukast blocks leukotriene receptors). Corticosteroids inhibit phospholipase A2 (via lipocortin), blocking both pathways.
| Eicosanoid | Source | Key Actions | Clinical Relevance |
|---|---|---|---|
| TXA2 (thromboxane) | Platelets (COX-1) | Platelet aggregation, vasoconstriction | Low-dose aspirin irreversibly inhibits COX-1 in platelets (cannot regenerate COX) → anti-thrombotic |
| PGI2 (prostacyclin) | Endothelium (COX-2) | Inhibits platelet aggregation, vasodilation | Opposes TXA2; endothelial cells regenerate COX → PGI2 production continues with low-dose aspirin |
| PGE2 | Many tissues | Vasodilation, fever, pain sensitization, gastric mucus/HCO3− secretion, maintains PDA | NSAIDs → ↓PGE2 → GI ulcers, premature PDA closure. Misoprostol (PGE analog) for NSAID ulcer prophylaxis |
| PGF2α | Uterus, eye | Uterine contraction, bronchoconstriction, ↓ intraocular pressure | Latanoprost (PGF analog) for glaucoma; PGF2α analogs for postpartum hemorrhage |
| LTB4 | Neutrophils | Neutrophil chemotaxis | Key mediator in acute inflammation |
| LTC4/D4/E4 | Mast cells, eosinophils | Bronchoconstriction, vascular permeability, mucus secretion | "Slow-reacting substances of anaphylaxis"; asthma treatment: montelukast, zileuton |
19 Lysosomal Storage Diseases
Lysosomal storage diseases are inherited deficiencies of lysosomal enzymes, leading to accumulation of undigested substrates within lysosomes. Most are autosomal recessive (except Fabry and Hunter, which are X-linked).
| Disease | Deficient Enzyme | Accumulated Substrate | Key Features |
|---|---|---|---|
| Tay-Sachs | Hexosaminidase A | GM2 ganglioside | Cherry-red macula, progressive neurodegeneration, no hepatosplenomegaly; Ashkenazi Jewish; "Tay-SaX = hexosaminidase" |
| Niemann-Pick (type A) | Sphingomyelinase | Sphingomyelin | Cherry-red macula, hepatosplenomegaly, progressive neurodegeneration; "No Man Picks his nose with his Sphinger" |
| Gaucher | Glucocerebrosidase (β-glucosidase) | Glucocerebroside | Hepatosplenomegaly, pancytopenia, bone crises, "crumpled tissue paper" macrophages (Gaucher cells); most common LSD |
| Fabry | α-Galactosidase A | Globotriaosylceramide (Gb3) | X-linked; peripheral neuropathy, angiokeratomas, renal failure, corneal dystrophy ("whorl") |
| Krabbe | Galactocerebrosidase | Galactocerebroside, psychosine | Peripheral neuropathy, optic atrophy, globoid cells |
| Metachromatic leukodystrophy | Arylsulfatase A | Sulfatides | Central & peripheral demyelination, ataxia, dementia |
| Hunter (MPS II) | Iduronate-2-sulfatase | Heparan sulfate, dermatan sulfate | X-linked; mild Hurler-like features; NO corneal clouding (vs. Hurler) |
| Hurler (MPS I) | α-L-iduronidase | Heparan sulfate, dermatan sulfate | Corneal clouding, coarse facies, hepatosplenomegaly, intellectual disability, gargoylism |
Cherry-red macula: Tay-Sachs and Niemann-Pick (also central retinal artery occlusion). Hepatosplenomegaly: Niemann-Pick, Gaucher (NOT Tay-Sachs). X-linked: Fabry and Hunter ("Fabry and the Hunter are X-men"). Enzyme replacement therapy (ERT) is available for Gaucher (imiglucerase), Fabry (agalsidase), Pompe (alglucosidase alfa), and several MPS types.
Sphingolipid Metabolism Overview
Sphingolipids are membrane lipids derived from ceramide (sphingosine + fatty acid). Ceramide synthesis begins with condensation of palmitoyl-CoA + serine by serine palmitoyltransferase. Adding phosphocholine yields sphingomyelin; adding sugars yields cerebrosides (one sugar) or gangliosides (oligosaccharide with sialic acid). Degradation occurs stepwise in lysosomes by specific hydrolases. Deficiency of any hydrolase causes accumulation of its substrate—the molecular basis of the sphingolipidoses listed above.
Sphingolipid Degradation Pathway
| Substrate | Enzyme | Product | Disease if Deficient |
|---|---|---|---|
| GM2 ganglioside | Hexosaminidase A | GM3 ganglioside | Tay-Sachs |
| Sphingomyelin | Sphingomyelinase | Ceramide | Niemann-Pick A/B |
| Glucocerebroside | Glucocerebrosidase | Ceramide | Gaucher |
| Globotriaosylceramide | α-Galactosidase A | Lactosylceramide | Fabry |
| Galactocerebroside | Galactocerebrosidase | Ceramide | Krabbe |
| Sulfatide | Arylsulfatase A | Galactocerebroside | Metachromatic leukodystrophy |
| Ceramide | Ceramidase | Sphingosine + FA | Farber disease |
I-Cell Disease (Mucolipidosis Type II)
I-cell disease results from deficiency of GlcNAc phosphotransferase, the enzyme that adds mannose-6-phosphate (M6P) tags to lysosomal enzymes in the Golgi. Without M6P, lysosomal enzymes are secreted extracellularly instead of being directed to lysosomes. Result: intracellular lysosomes lack enzymes and fill with undigested substrates (dense "inclusion bodies" → "I-cell"). Clinically resembles severe Hurler syndrome: coarse facies, skeletal abnormalities, restricted joint mobility, psychomotor retardation. Serum lysosomal enzyme levels are elevated (misdirected into blood).
20 Amino Acid Metabolism & Inborn Errors
Amino acid catabolism involves removal of the α-amino group (via transamination to α-ketoglutarate forming glutamate, then oxidative deamination by glutamate dehydrogenase releasing NH4+) and disposal of the carbon skeleton. Ammonia is detoxified by the urea cycle in the liver.
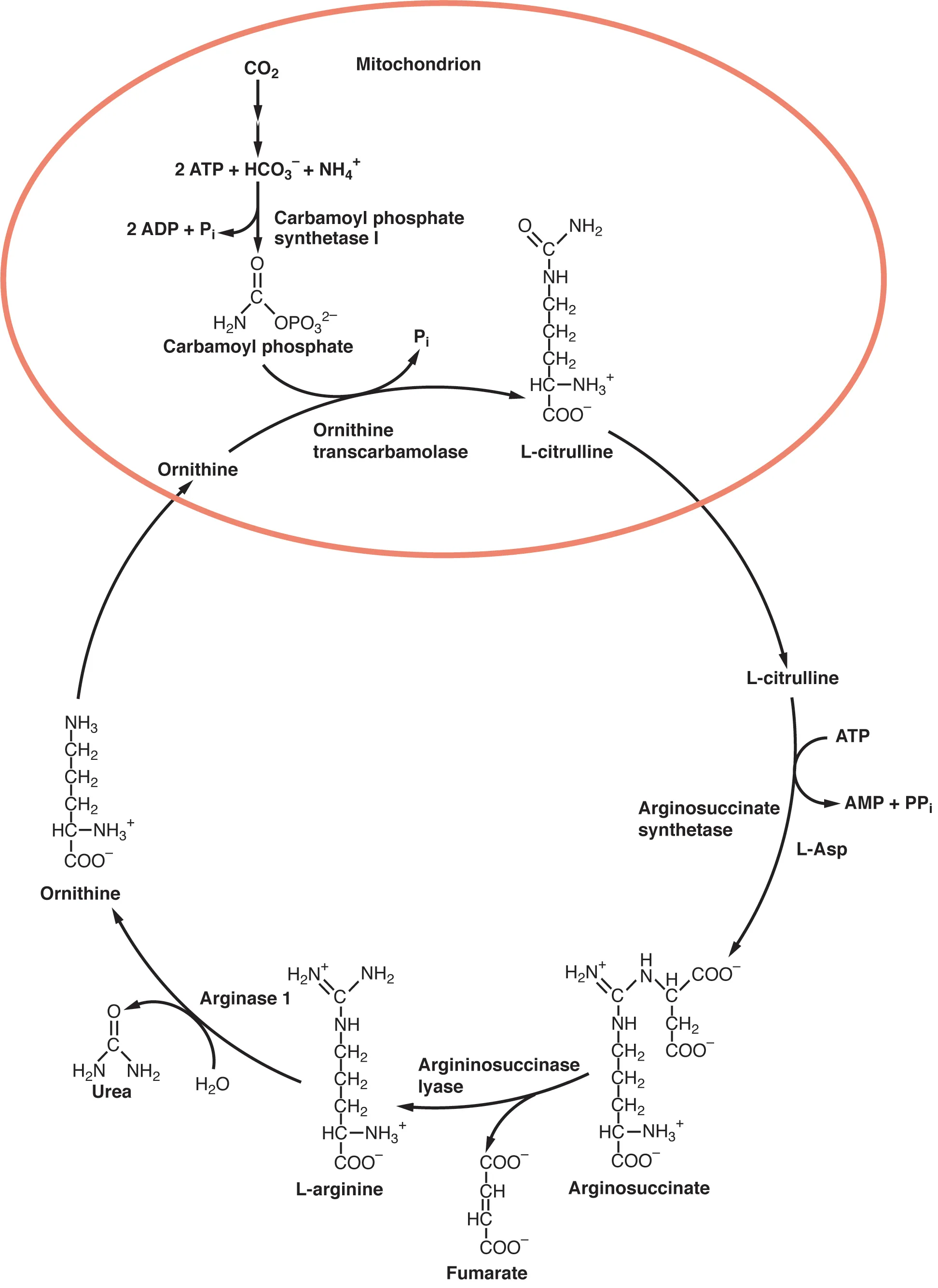
Urea Cycle
The urea cycle converts two nitrogen atoms (one from NH4+ via carbamoyl phosphate, one from aspartate) and CO2 into urea for renal excretion. The cycle spans two compartments: the first two steps (CPS I and OTC) occur in the mitochondrial matrix; the remaining three steps (argininosuccinate synthetase, argininosuccinate lyase, arginase) occur in the cytoplasm.
| Enzyme | Location | Reaction | Notes |
|---|---|---|---|
| CPS I (rate-limiting) | Mitochondria | NH4+ + CO2 + 2 ATP → carbamoyl phosphate | Activated by N-acetylglutamate (NAG); distinct from CPS II (pyrimidine synthesis, cytoplasmic) |
| OTC | Mitochondria | Carbamoyl phosphate + ornithine → citrulline | Most common urea cycle defect (X-linked); ↑orotic acid (CP shunted to pyrimidines) |
| Argininosuccinate synthetase | Cytoplasm | Citrulline + aspartate → argininosuccinate | Aspartate donates the 2nd nitrogen atom; deficiency = citrullinemia type I |
| Argininosuccinate lyase | Cytoplasm | Argininosuccinate → arginine + fumarate | Fumarate links urea cycle to TCA cycle (aspartate-argininosuccinate shunt) |
| Arginase | Cytoplasm | Arginine → urea + ornithine | Ornithine is recycled back to mitochondria; urea excreted by kidneys |
Defects in any urea cycle enzyme cause hyperammonemia with lethargy, vomiting, cerebral edema, and respiratory alkalosis (NH3 stimulates respiratory center). Treatment: low-protein diet, nitrogen scavengers (sodium benzoate conjugates glycine, sodium phenylbutyrate conjugates glutamine), lactulose (promotes NH3 excretion by colonic bacteria), dialysis in acute crisis.
Inborn Errors of Amino Acid Metabolism
| Disease | Deficient Enzyme | Accumulated | Key Features |
|---|---|---|---|
| Phenylketonuria (PKU) | Phenylalanine hydroxylase (or BH4 cofactor) | Phenylalanine, phenylketones | Intellectual disability, fair skin/hair, musty body odor, eczema; detected on newborn screen; Tx: low-Phe diet, BH4 |
| Maple syrup urine disease | Branched-chain α-ketoacid dehydrogenase | BCAAs (Ile, Leu, Val) & α-keto acids | Sweet-smelling urine, intellectual disability, feeding difficulty; Tx: restrict BCAAs, thiamine trial |
| Homocystinuria | Cystathionine β-synthase (most common) or methionine synthase | Homocysteine, methionine | Marfanoid habitus, lens subluxation (downward), intellectual disability, thromboembolism, osteoporosis; Tx: B6, B12, folate, restrict Met |
| Cystinuria | Renal PCT transporter (dibasic AAs) | Cystine in urine | Recurrent kidney stones (cystine = hexagonal crystals); Tx: hydration, alkalinize urine, penicillamine |
| Alkaptonuria | Homogentisic acid oxidase | Homogentisic acid | Dark urine on standing, ochronosis (blue-black pigmentation of cartilage), early arthritis |
| Hartnup disease | Neutral amino acid transporter (gut/kidney) | Tryptophan malabsorption | Pellagra-like symptoms (tryptophan needed for niacin synthesis); neutral aminoaciduria |
Both present with tall stature and long limbs. Key differentiators: Lens subluxation direction: downward in homocystinuria, upward in Marfan. Thromboembolism: present in homocystinuria, not in Marfan. Intellectual disability: present in homocystinuria, not in Marfan. Inheritance: AR in homocystinuria, AD in Marfan.
Heme Synthesis & Porphyrias
Heme synthesis begins in the mitochondria (ALA synthase: rate-limiting, requires B6, inhibited by heme) and continues in the cytoplasm before returning to mitochondria for the final step (ferrochelatase inserts Fe2+ into protoporphyrin IX). The porphyrias are enzyme deficiencies in the heme synthesis pathway.
| Porphyria | Deficient Enzyme | Accumulated | Key Features |
|---|---|---|---|
| Acute intermittent porphyria (AIP) | Porphobilinogen deaminase (HMB synthase) | PBG, ALA (urine) | AD; abdominal pain, neuropsychiatric symptoms, port-wine colored urine; NO photosensitivity. Precipitated by drugs (P450 inducers), fasting, alcohol. Tx: hemin, glucose loading. |
| Porphyria cutanea tarda (PCT) | Uroporphyrinogen decarboxylase | Uroporphyrin | Most common porphyria; photosensitivity (blistering skin on sun-exposed areas), hypertrichosis, tea-colored urine. Associated with hepatitis C, alcohol, iron overload. Tx: phlebotomy, hydroxychloroquine. |
| Erythropoietic protoporphyria | Ferrochelatase | Protoporphyrin IX | Painful photosensitivity in childhood (immediate burning, not blistering); elevated free erythrocyte protoporphyrin. |
| Lead poisoning | Inhibits ALA dehydratase & ferrochelatase | ALA, protoporphyrin IX | Mimics porphyria; basophilic stippling, microcytic anemia, abdominal colic, lead lines on gums/bones, wrist drop, encephalopathy in children |
21 Purine & Pyrimidine Metabolism
Purines (adenine, guanine) are synthesized de novo on a ribose-5-phosphate scaffold (from the PPP) using amino acids (Gly, Asp, Gln), CO2, and THF derivatives. IMP is the first purine nucleotide formed. Pyrimidines (cytosine, uracil, thymine) are built as a free base first (orotate), then attached to ribose-5-phosphate.
Key Enzymes & Pharmacological Targets
| Enzyme | Pathway | Relevance |
|---|---|---|
| PRPP synthetase | Purine & pyrimidine synthesis | Overactivity → gout (↑ uric acid) |
| HGPRT (hypoxanthine-guanine phosphoribosyltransferase) | Purine salvage | Deficiency → Lesch-Nyhan syndrome |
| Xanthine oxidase | Purine degradation | Converts hypoxanthine → xanthine → uric acid; target of allopurinol |
| Dihydrofolate reductase (DHFR) | THF regeneration (purine & pyrimidine synthesis) | Target of methotrexate, trimethoprim, pyrimethamine |
| Thymidylate synthase | dTMP synthesis | Target of 5-fluorouracil (5-FU) |
| Ribonucleotide reductase | dNTP synthesis (DNA) | Target of hydroxyurea (also induces HbF) |
| Dihydroorotate dehydrogenase | Pyrimidine synthesis | Target of leflunomide (RA treatment) |
Gout & Lesch-Nyhan Syndrome
Gout: hyperuricemia → monosodium urate crystal deposition in joints, causing acute inflammatory arthritis. Negatively birefringent, needle-shaped crystals under polarized light. Treatment: acute (NSAIDs, colchicine, steroids); chronic (allopurinol [xanthine oxidase inhibitor], febuxostat, probenecid [uricosuric]).
Lesch-Nyhan syndrome: X-linked deficiency of HGPRT → excess purine synthesis (cannot salvage) → severe hyperuricemia, gout, intellectual disability, self-mutilating behavior (lip and finger biting), choreoathetosis. Mnemonic: "HGPRT—He's Got Purine Recovery Trouble."
Adenosine Deaminase (ADA) Deficiency
ADA deficiency causes severe combined immunodeficiency (SCID). ADA normally converts adenosine and deoxyadenosine to inosine and deoxyinosine. When ADA is absent, deoxyadenosine accumulates and is phosphorylated to dATP, which inhibits ribonucleotide reductase. This prevents DNA synthesis, particularly devastating to lymphocytes (which rely heavily on de novo nucleotide synthesis). Result: absent T cells and B cells → life-threatening infections from infancy. Treatment: enzyme replacement (PEG-ADA), hematopoietic stem cell transplant, or gene therapy (one of the first successful gene therapy targets).
Pseudogout vs. Gout
| Feature | Gout (Urate) | Pseudogout (CPPD) |
|---|---|---|
| Crystal | Monosodium urate | Calcium pyrophosphate dihydrate (CPPD) |
| Shape | Needle-shaped | Rhomboid-shaped |
| Birefringence | Strongly negatively birefringent (yellow when parallel to polarizer) | Weakly positively birefringent (blue when parallel) |
| Joint(s) | 1st MTP (podagra), ankles, knees | Knee (most common), wrists |
| X-ray | Tophi, "punched-out" erosions with overhanging edges | Chondrocalcinosis (calcification of articular cartilage) |
| Associations | Hyperuricemia, alcohol, purine-rich diet, thiazides, tumor lysis | Hyperparathyroidism, hemochromatosis, hypomagnesemia, hypothyroidism |
Purine vs. Pyrimidine Synthesis Comparison
| Feature | Purine Synthesis | Pyrimidine Synthesis |
|---|---|---|
| Ring assembly | Built on ribose-5-phosphate scaffold (PRPP) | Ring assembled first (as orotate), then attached to ribose |
| First committed step enzyme | Glutamine-PRPP amidotransferase | CPS II (cytoplasmic; distinct from CPS I of urea cycle) |
| Atoms contributed by | Gly, Asp, Gln, CO2, N10-formyl-THF | Gln, Asp, CO2 |
| Feedback inhibition | AMP, GMP, IMP inhibit amidotransferase | UTP inhibits CPS II |
| Salvage pathway | HGPRT (hypoxanthine/guanine), APRT (adenine) | Thymidine kinase, uridine kinase |
| Degradation end product | Uric acid (by xanthine oxidase) | CO2, NH3, β-alanine (water-soluble, readily excreted) |
Chemotherapy Targeting Nucleotide Metabolism
| Drug | Target | Mechanism | Clinical Use |
|---|---|---|---|
| Methotrexate | DHFR (dihydrofolate reductase) | Inhibits THF regeneration → ↓dTMP and purine synthesis | Leukemia, RA, ectopic pregnancy; rescued with leucovorin (folinic acid) |
| 5-Fluorouracil (5-FU) | Thymidylate synthase | 5-FdUMP irreversibly inhibits dTMP synthesis | Colorectal, breast, head/neck cancers |
| 6-Mercaptopurine (6-MP) | Purine synthesis (multiple) | Purine analog blocks de novo synthesis | ALL; metabolized by TPMT (check genotype) and xanthine oxidase (dose reduce with allopurinol) |
| Hydroxyurea | Ribonucleotide reductase | Inhibits conversion of NDP → dNDP | CML, sickle cell disease (also induces HbF); myelosuppressive |
| Mycophenolate | IMP dehydrogenase | Blocks GMP synthesis → selective lymphocyte inhibition | Transplant rejection prophylaxis, lupus nephritis |
22 Fat-Soluble Vitamins (A, D, E, K)
Fat-soluble vitamins are absorbed with dietary fat via micelles, transported in chylomicrons, and stored in liver and adipose tissue. Deficiency may result from fat malabsorption (e.g., cystic fibrosis, celiac disease, cholestasis, short bowel syndrome, pancreatic insufficiency).
| Vitamin | Active Form / Function | Deficiency | Toxicity |
|---|---|---|---|
| A (retinol) | Retinal (vision), retinoic acid (gene expression, differentiation), retinol (antioxidant). Component of rhodopsin in rod cells. | Night blindness (nyctalopia), Bitot spots (conjunctival keratinization), xerophthalmia, dry skin, immune suppression, keratomalacia | Pseudotumor cerebri (IIH), hepatotoxicity, teratogenesis (isotretinoin), skin peeling |
| D (cholecalciferol) | 1,25-(OH)2D3 (calcitriol): increases intestinal Ca2+ & PO43− absorption, promotes bone mineralization. Synthesized in skin (UV-B), hydroxylated in liver (25-OH) then kidney (1,25-OH). | Rickets (children): bowed legs, craniotabes, rachitic rosary. Osteomalacia (adults): bone pain, proximal myopathy, ↓Ca2+, ↓PO4, ↑ALP, ↑PTH | Hypercalcemia: nausea, polyuria, nephrocalcinosis, metastatic calcification |
| E (tocopherol) | Antioxidant (protects cell membranes from lipid peroxidation by scavenging free radicals) | Hemolytic anemia (especially premature infants), posterior column & spinocerebellar degeneration (ataxia), peripheral neuropathy | May increase bleeding risk (potentiates warfarin) |
| K (phylloquinone/menaquinone) | Cofactor for γ-carboxylation of glutamate residues on clotting factors II, VII, IX, X, protein C & S. Activated by vitamin K epoxide reductase (target of warfarin). | Bleeding diathesis: ↑PT/INR. Hemorrhagic disease of the newborn (neonatal vitamin K deficiency). | Hemolytic anemia in neonates (if given excess menadione/K3) |
Fat-Soluble Vitamin Absorption
Fat-soluble vitamins (A, D, E, K) require bile salts for micelle formation and intestinal absorption. Any condition causing fat malabsorption will lead to deficiency of all four vitamins: cystic fibrosis (pancreatic insufficiency), celiac disease (villous atrophy), Crohn disease (terminal ileum), cholestatic liver disease (bile flow obstruction), short bowel syndrome, and chronic pancreatitis. Patients with these conditions require supplementation and monitoring of fat-soluble vitamin levels.
Vitamin A & Retinoid Signaling
Vitamin A exists in three active forms: retinal (component of rhodopsin in rod cells, essential for dark adaptation), retinoic acid (binds nuclear receptors RAR/RXR to regulate gene expression, cell differentiation, and immune function), and retinol (transport and storage form). Retinoic acid is critical for epithelial integrity and immune function. Its role in cell differentiation is exploited therapeutically: all-trans retinoic acid (ATRA) is used to treat acute promyelocytic leukemia (APL; PML-RARa fusion), inducing differentiation of promyelocytes. Isotretinoin (13-cis-retinoic acid) is used for severe acne but is a potent teratogen (iPLEDGE program required).
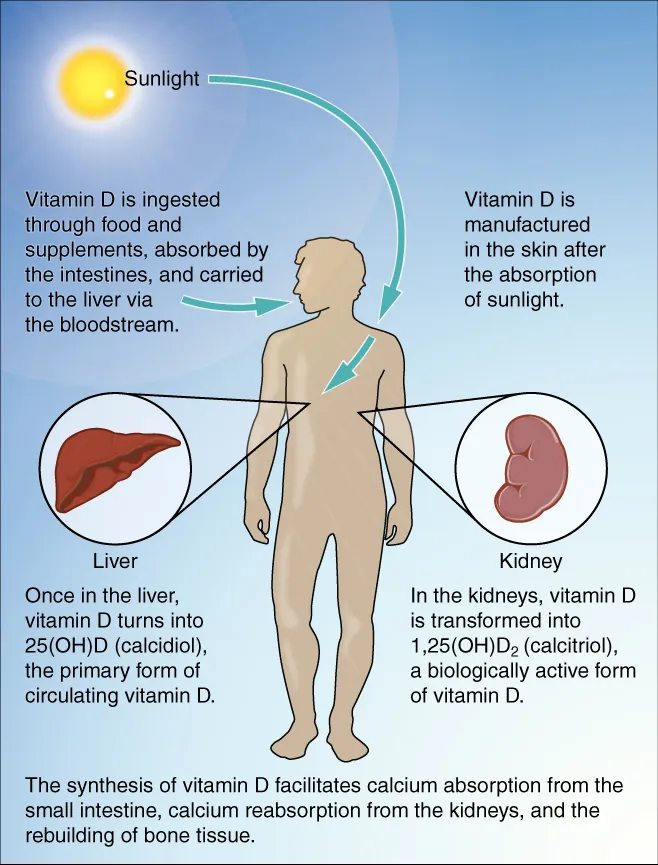
Vitamin D Metabolism in Detail
The vitamin D pathway: (1) 7-dehydrocholesterol in skin is converted to cholecalciferol (D3) by UV-B radiation. (2) Liver 25-hydroxylase produces 25-OH-D3 (calcidiol)—the best clinical measure of vitamin D status. (3) Kidney 1α-hydroxylase produces 1,25-(OH)2D3 (calcitriol)—the most active form. 1α-Hydroxylase is stimulated by PTH and low phosphate, and inhibited by FGF-23 and high calcium/phosphate. Calcitriol acts on intestine (↑Ca2+ and PO4 absorption), bone (mobilization of Ca2+/PO4), and kidney (↑Ca2+ reabsorption). Vitamin D2 (ergocalciferol) is plant-derived and undergoes the same hydroxylation steps.
23 Water-Soluble Vitamins (B Complex & C)
Water-soluble vitamins generally serve as enzyme cofactors. They are not stored extensively (except B12 in liver, lasting 3–5 years), so deficiencies develop more rapidly than fat-soluble vitamin deficiencies.
| Vitamin | Cofactor Form / Function | Deficiency Syndrome |
|---|---|---|
| B1 (thiamine) | TPP: cofactor for pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, transketolase (PPP), branched-chain α-ketoacid dehydrogenase | Beriberi: wet (high-output cardiac failure, edema) or dry (peripheral neuropathy, muscle wasting). Wernicke-Korsakoff: confusion, ataxia, ophthalmoplegia (Wernicke); confabulation, memory loss (Korsakoff). Common in alcoholism. |
| B2 (riboflavin) | FAD, FMN: cofactors in redox reactions (ETC Complex I & II, fatty acid oxidation) | Cheilosis (cracking at corners of mouth), glossitis, corneal vascularization, normocytic anemia |
| B3 (niacin) | NAD+, NADP+: redox coenzymes. Derived from tryptophan (requires B2, B6). | Pellagra: 3 D's — Diarrhea, Dermatitis (sun-exposed, Casal necklace), Dementia (+ Death if untreated). Seen in alcoholism, Hartnup disease, carcinoid syndrome, isoniazid use. |
| B5 (pantothenic acid) | CoA, fatty acid synthase (ACP): essential for TCA cycle, fatty acid metabolism | Rare; dermatitis, enteritis, alopecia, adrenal insufficiency ("burning feet syndrome") |
| B6 (pyridoxine) | PLP: cofactor for transamination, decarboxylation, heme synthesis (ALA synthase), neurotransmitter synthesis (serotonin, dopamine, GABA, histamine), cystathionine synthase | Peripheral neuropathy, sideroblastic anemia (impaired heme synthesis), seizures (decreased GABA). Caused by isoniazid (INH binds PLP). Tx: supplement B6 with INH. |
| B7 (biotin) | Cofactor for carboxylases: pyruvate carboxylase, acetyl-CoA carboxylase, propionyl-CoA carboxylase | Dermatitis, alopecia, enteritis. Caused by excessive raw egg white consumption (avidin binds biotin) or prolonged antibiotics. |
| B9 (folate) | THF: one-carbon carrier for purine synthesis, dTMP synthesis (thymidylate synthase), methionine regeneration | Megaloblastic anemia (hypersegmented neutrophils, macrocytic RBCs), neural tube defects in pregnancy. Deficiency in first months of restricted diet (no hepatic stores like B12). |
| B12 (cobalamin) | Cofactor for methionine synthase (homocysteine → methionine; requires B9) and methylmalonyl-CoA mutase | Megaloblastic anemia + neurological symptoms (subacute combined degeneration: posterior columns, lateral corticospinal tracts, peripheral neuropathy). ↑ Homocysteine AND ↑ methylmalonic acid (distinguishes from folate deficiency). Causes: pernicious anemia, vegan diet, Diphyllobothrium latum, gastrectomy, Crohn terminal ileum. |
| C (ascorbic acid) | Cofactor for prolyl & lysyl hydroxylase (collagen synthesis), dopamine β-hydroxylase (NE synthesis), iron absorption (Fe3+ → Fe2+) | Scurvy: swollen bleeding gums, perifollicular hemorrhage, petechiae, poor wound healing, corkscrew hairs, subperiosteal hemorrhage, anemia. Weakness reflects impaired collagen & carnitine synthesis. |
Both cause megaloblastic anemia with hypersegmented neutrophils. Both elevate homocysteine. Only B12 deficiency elevates methylmalonic acid (MMA) and causes neurological damage (subacute combined degeneration). Administering folate to a patient with undiagnosed B12 deficiency can correct the anemia but will NOT prevent neurological deterioration—making it critical to check B12 before treating with folate alone.
Thiamine (B1) Deficiency in Detail
Thiamine pyrophosphate (TPP) is the cofactor for four critical enzyme complexes: pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, branched-chain α-ketoacid dehydrogenase, and transketolase (PPP). Alcoholism is the most common cause of B1 deficiency in developed countries (poor intake, impaired absorption, increased demand). Wernicke encephalopathy is a medical emergency: classic triad of confusion, ophthalmoplegia (CN VI palsy), and ataxia (cerebellar). If untreated, it progresses to Korsakoff syndrome: irreversible anterograde amnesia with confabulation (mammillary body and dorsomedial thalamic nuclei damage). Treatment: IV thiamine BEFORE glucose administration (glucose metabolism consumes B1 and can precipitate Wernicke in a thiamine-depleted patient).
Niacin (B3) Pharmacology
At pharmacological doses, niacin lowers triglycerides and LDL while raising HDL (most effective HDL-raising agent). It inhibits lipolysis in adipose tissue (reducing hepatic VLDL production). Major side effect: flushing (prostaglandin-mediated cutaneous vasodilation); reduced by aspirin pretreatment or extended-release formulation. Other adverse effects: hyperglycemia, hyperuricemia, hepatotoxicity.
Folate & Neural Tube Defects
Folate supplementation (400 μg/day) beginning at least one month before conception and continuing through the first trimester dramatically reduces the incidence of neural tube defects (anencephaly, spina bifida). Women with a prior NTD-affected pregnancy should take 4 mg/day. Folate fortification of grain products (mandatory in the US since 1998) has reduced NTD incidence by ~25–30%. Methotrexate and other antifolates are teratogenic and absolutely contraindicated in pregnancy.
Vitamin Toxicity Quick-Reference
| Vitamin | Toxicity Findings | Mechanism |
|---|---|---|
| A (hypervitaminosis A) | Pseudotumor cerebri (headache, papilledema), hepatotoxicity, skin desquamation, teratogenesis | Retinoic acid excess disrupts cell differentiation; fat-soluble → accumulates |
| D | Hypercalcemia: nausea, vomiting, polyuria, constipation, renal stones, nephrocalcinosis, metastatic calcification | Excess calcitriol ↑Ca2+ absorption and bone resorption |
| E | Increased bleeding risk, potentiation of warfarin | Inhibits vitamin K-dependent carboxylation at high doses |
| B6 (megadose) | Peripheral sensory neuropathy (ataxia, loss of proprioception) | Direct neurotoxicity at doses >200 mg/day chronically |
| B3 (niacin) | Flushing, hyperglycemia, hyperuricemia, hepatotoxicity | Prostaglandin-mediated vasodilation; metabolic effects at pharmacological doses |
| C (megadose) | Nausea, diarrhea, increased oxalate kidney stones, false-negative guaiac test | Excess oxalate production; reducing agent interferes with tests |
24 Minerals, Trace Elements & Nutrition
Minerals and trace elements serve as enzyme cofactors, structural components, and electrolytes essential for physiological function.
Key Minerals & Trace Elements
| Element | Function | Deficiency | Excess / Notes |
|---|---|---|---|
| Iron (Fe) | Heme (hemoglobin, myoglobin, cytochromes), FeS clusters in ETC | Microcytic hypochromic anemia, pica, koilonychia, ↓ferritin, ↑TIBC | Hemochromatosis (HFE mutation): bronze skin, cirrhosis, DM, cardiomyopathy, arthropathy |
| Copper (Cu) | Lysyl oxidase, ceruloplasmin, cytochrome c oxidase, SOD, dopamine β-hydroxylase, tyrosinase | Menkes disease (X-linked Cu transport defect): kinky hair, connective tissue laxity, neurodegeneration | Wilson disease (AR, ATP7B mutation): Kayser-Fleischer rings, hepatolenticular degeneration, psychiatric symptoms, low ceruloplasmin |
| Zinc (Zn) | Metalloenzymes (carbonic anhydrase, alcohol dehydrogenase, collagenase), wound healing, immunity, taste/smell | Acrodermatitis enteropathica (AR), delayed wound healing, hypogeusia, anosmia, impaired immunity, hypogonadism | Can cause copper deficiency (zinc induces metallothionein which sequesters Cu) |
| Selenium (Se) | Glutathione peroxidase, thioredoxin reductase, deiodinases | Keshan disease (cardiomyopathy), Kashin-Beck disease (osteoarthropathy) | Garlic breath, hair/nail loss, GI upset |
| Chromium (Cr) | Glucose tolerance factor (potentiates insulin) | Impaired glucose tolerance, peripheral neuropathy | Rare toxicity |
| Fluoride (F) | Incorporated into hydroxyapatite (fluoroapatite) strengthening enamel | Dental caries | Fluorosis (mottled enamel, skeletal fluorosis) |
Nutrition Disorders
Kwashiorkor: protein malnutrition with adequate caloric intake. Features: edema (low albumin → low oncotic pressure), fatty liver, skin lesions, flag sign (alternating pigmented/depigmented hair bands), preserved fat stores. Marasmus: total calorie deficiency. Features: muscle wasting, loss of subcutaneous fat, emaciation, no edema. Refeeding syndrome: occurs when malnourished patients are fed too quickly. Insulin surge drives PO43−, K+, and Mg2+ into cells → dangerous hypophosphatemia → cardiac arrhythmias, respiratory failure, rhabdomyolysis.
Iron Metabolism
Dietary iron is absorbed in the duodenum as heme iron (from meat, directly absorbed) or non-heme ferric iron (Fe3+, reduced to Fe2+ by duodenal cytochrome B, then transported by DMT-1). Inside enterocytes, iron is either stored as ferritin or exported by ferroportin (the only known iron exporter). In blood, iron is bound to transferrin and delivered to cells via transferrin receptor. Hepcidin, produced by the liver, is the master regulator: it degrades ferroportin, reducing iron absorption and release from macrophages. Hepcidin is upregulated by iron excess and inflammation (explaining anemia of chronic disease) and downregulated by iron deficiency, erythropoietic activity, and hypoxia.
| Condition | Ferritin | Serum Iron | TIBC | Transferrin Sat | Hepcidin |
|---|---|---|---|---|---|
| Iron deficiency anemia | ↓ | ↓ | ↑ | ↓ | ↓ |
| Anemia of chronic disease | ↑ (or normal) | ↓ | ↓ | ↓ | ↑ |
| Hemochromatosis | ↑↑ | ↑ | ↓ | ↑↑ | ↓ |
| Sideroblastic anemia | ↑ | ↑ | ↓ | ↑ | Variable |
| Pregnancy | ↓ | ↓ | ↑ | ↓ | ↓ |
Wilson Disease vs. Hemochromatosis
| Feature | Wilson Disease (Copper) | Hemochromatosis (Iron) |
|---|---|---|
| Inheritance | AR (ATP7B gene, chr 13) | AR (HFE gene, chr 6; C282Y mutation) |
| Pathophysiology | Impaired biliary copper excretion → copper accumulation | Excessive iron absorption (low hepcidin) → iron deposition |
| Liver | Hepatitis, cirrhosis, acute liver failure | Cirrhosis, hepatocellular carcinoma |
| Neuropsychiatric | Tremor, dysarthria, personality change, depression | Uncommon |
| Pathognomonic sign | Kayser-Fleischer rings (copper in Descemet membrane) | Bronze skin pigmentation |
| Lab findings | ↓ Ceruloplasmin, ↑ urine copper, ↑ hepatic copper | ↑↑ Ferritin, ↑ transferrin saturation, HFE genotyping |
| Other organs | Hemolytic anemia (Coombs-negative), renal tubular defects | Diabetes ("bronze diabetes"), cardiomyopathy, arthropathy, hypogonadism |
| Treatment | Penicillamine or trientine (copper chelation), zinc (blocks absorption) | Phlebotomy (first-line), deferoxamine (iron chelation) |
25 Molecular Biology & DNA Repair
The central dogma of molecular biology describes the flow of genetic information: DNA → RNA (transcription) → Protein (translation). Understanding these processes is essential for interpreting molecular diagnostics and understanding antibiotic and chemotherapeutic mechanisms.
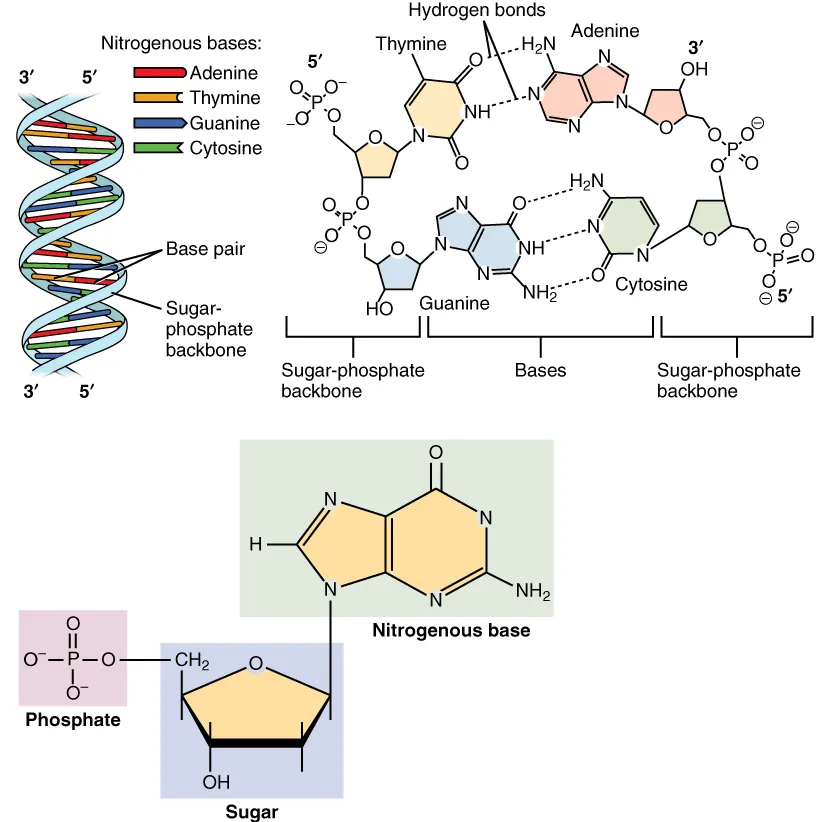
DNA Replication
Replication is semiconservative, bidirectional, and proceeds 5′ → 3′. Helicase unwinds the double helix. Topoisomerase relieves supercoiling (fluoroquinolones inhibit bacterial topoisomerase II/DNA gyrase). Primase synthesizes RNA primers. DNA polymerase III (prokaryotes) / Pol δ and Pol ε (eukaryotes) elongate the new strand. Leading strand is synthesized continuously; lagging strand is synthesized as Okazaki fragments, joined by DNA ligase. Telomerase (a reverse transcriptase) maintains telomere length in germ cells, stem cells, and most cancer cells.
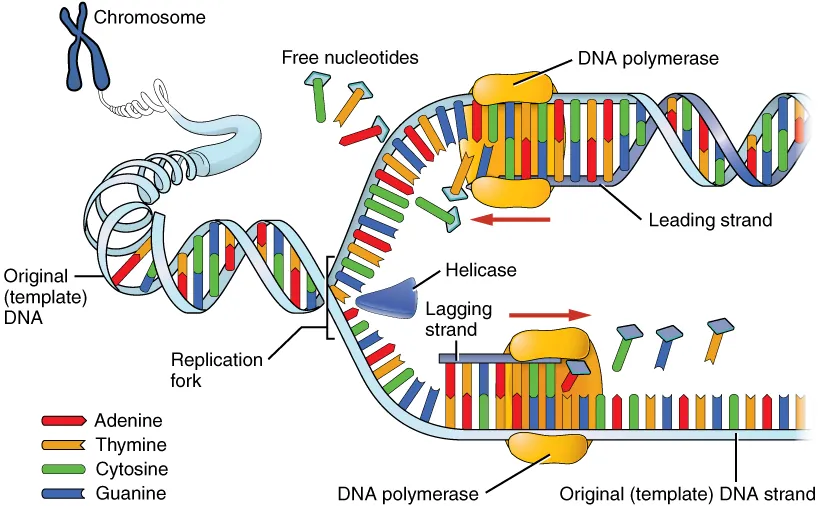
DNA Repair Mechanisms
| Repair Mechanism | Type of Damage | Key Features | Disease if Defective |
|---|---|---|---|
| Nucleotide excision repair (NER) | Bulky DNA adducts, thymine dimers (UV damage) | Excises oligonucleotide containing lesion | Xeroderma pigmentosum (extreme UV sensitivity, skin cancers) |
| Base excision repair (BER) | Small base modifications (deamination, oxidation, methylation) | Glycosylase removes damaged base → AP endonuclease → gap filled | Rare; MUTYH-associated polyposis |
| Mismatch repair (MMR) | Mismatched bases from replication errors | MSH2/MLH1 recognize mismatches on new strand | Lynch syndrome (hereditary nonpolyposis colorectal cancer, HNPCC) |
| Homologous recombination | Double-strand breaks | Uses sister chromatid as template; error-free | BRCA1/BRCA2 mutations → breast/ovarian cancer |
| Non-homologous end joining (NHEJ) | Double-strand breaks | Joins broken ends directly; error-prone | May introduce mutations; used throughout cell cycle |
Key Antibiotics & Their Molecular Targets
| Target | Drug(s) | Mechanism |
|---|---|---|
| DNA gyrase / Topoisomerase IV | Fluoroquinolones (ciprofloxacin, levofloxacin) | Prevent supercoil relaxation → block replication |
| RNA polymerase | Rifampin | Inhibits bacterial DNA-dependent RNA polymerase |
| 30S ribosomal subunit | Aminoglycosides, tetracyclines | Aminoglycosides: misread mRNA; Tetracyclines: block tRNA binding |
| 50S ribosomal subunit | Macrolides, chloramphenicol, clindamycin, linezolid | Block translocation or peptidyl transfer |
| Folate synthesis | Sulfonamides (DHPS), Trimethoprim (DHFR) | Block sequential steps in folate pathway |
Transcription
RNA polymerase synthesizes RNA 5′ → 3′ from a DNA template (read 3′ → 5′). Eukaryotes have three RNA polymerases: Pol I (rRNA, except 5S), Pol II (mRNA, snRNA, miRNA), Pol III (tRNA, 5S rRNA). α-Amanitin (Amanita phalloides mushroom toxin) inhibits RNA Pol II → hepatotoxicity. Eukaryotic mRNA is processed: 5′ cap (7-methylguanosine, added by guanylyltransferase), 3′ poly-A tail (added by poly-A polymerase), and splicing (introns removed by the spliceosome). The 5′ cap is critical for ribosome recognition and mRNA stability.
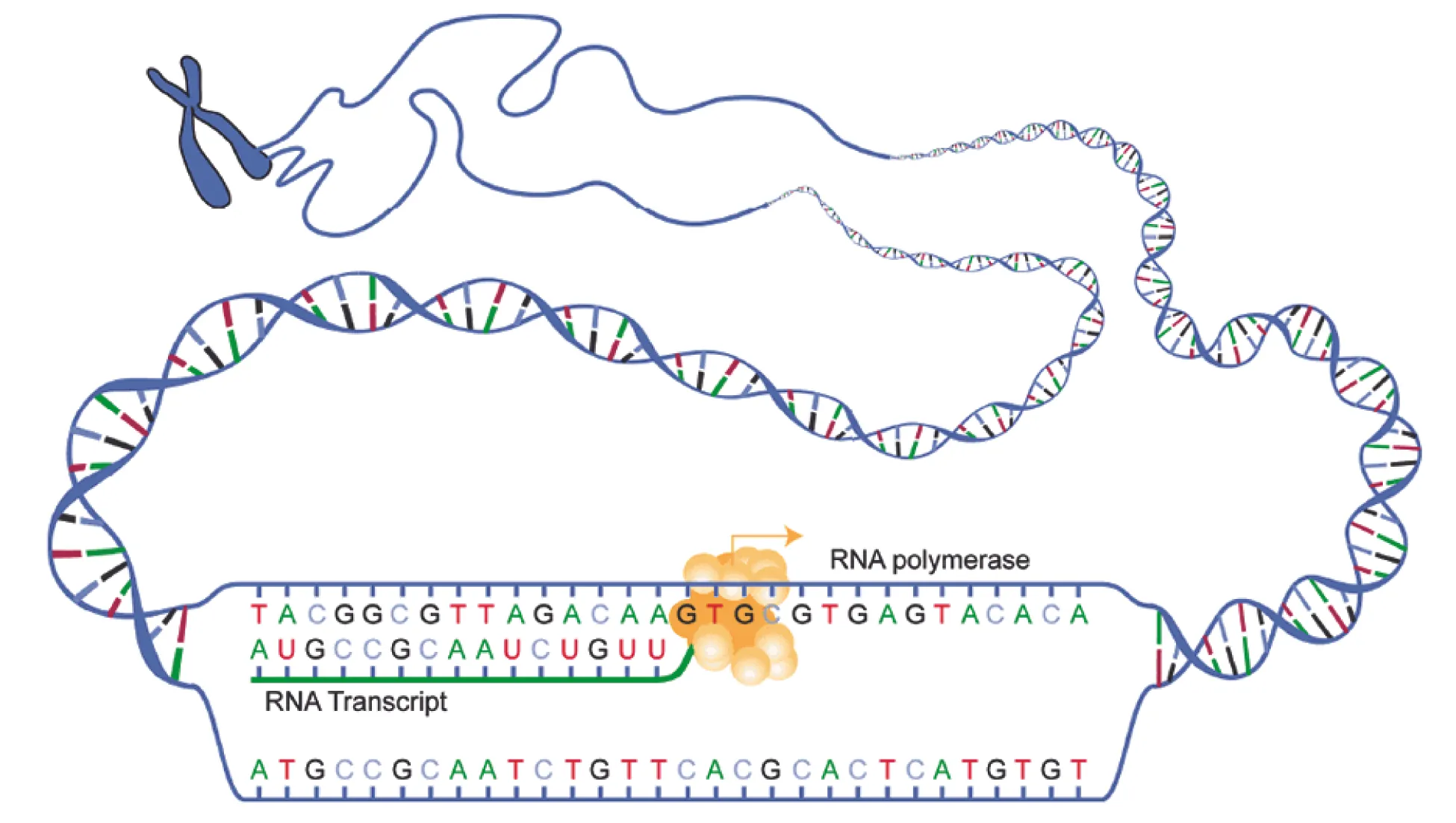
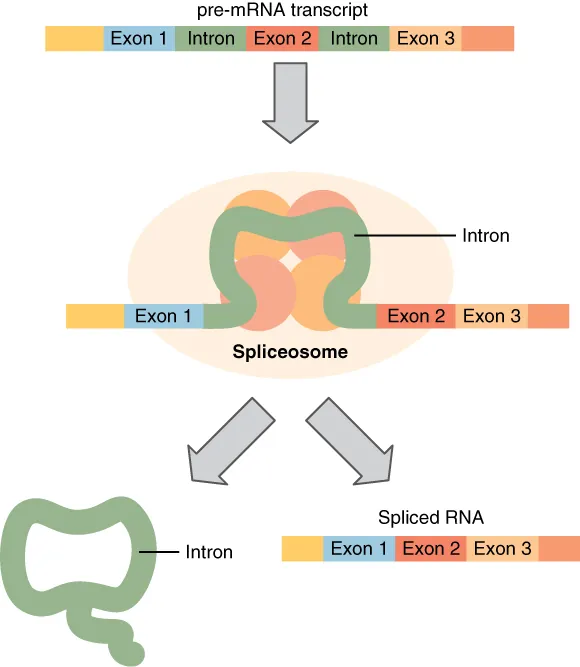
Translation
Translation occurs on ribosomes (80S in eukaryotes = 40S + 60S; 70S in prokaryotes = 30S + 50S). The process involves three stages: Initiation (small subunit binds mRNA, start codon AUG recognized, Met-tRNA loaded), Elongation (aminoacyl-tRNA binds A site, peptidyl transferase forms peptide bond, ribosome translocates), and Termination (stop codons UAA, UAG, UGA; release factors). Diphtheria toxin inactivates EF-2 (elongation factor 2) via ADP-ribosylation. Pseudomonas exotoxin A has the same mechanism.
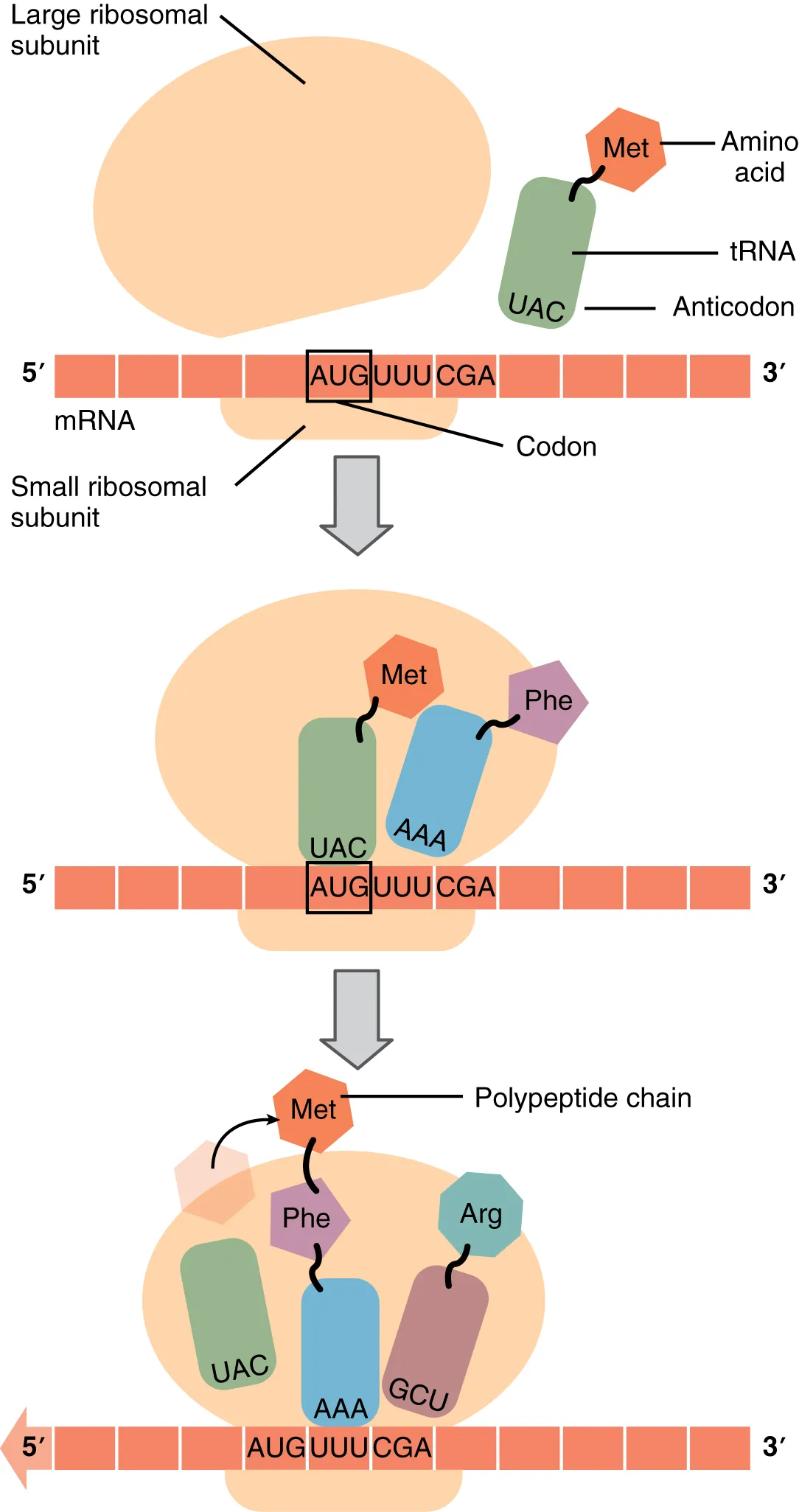
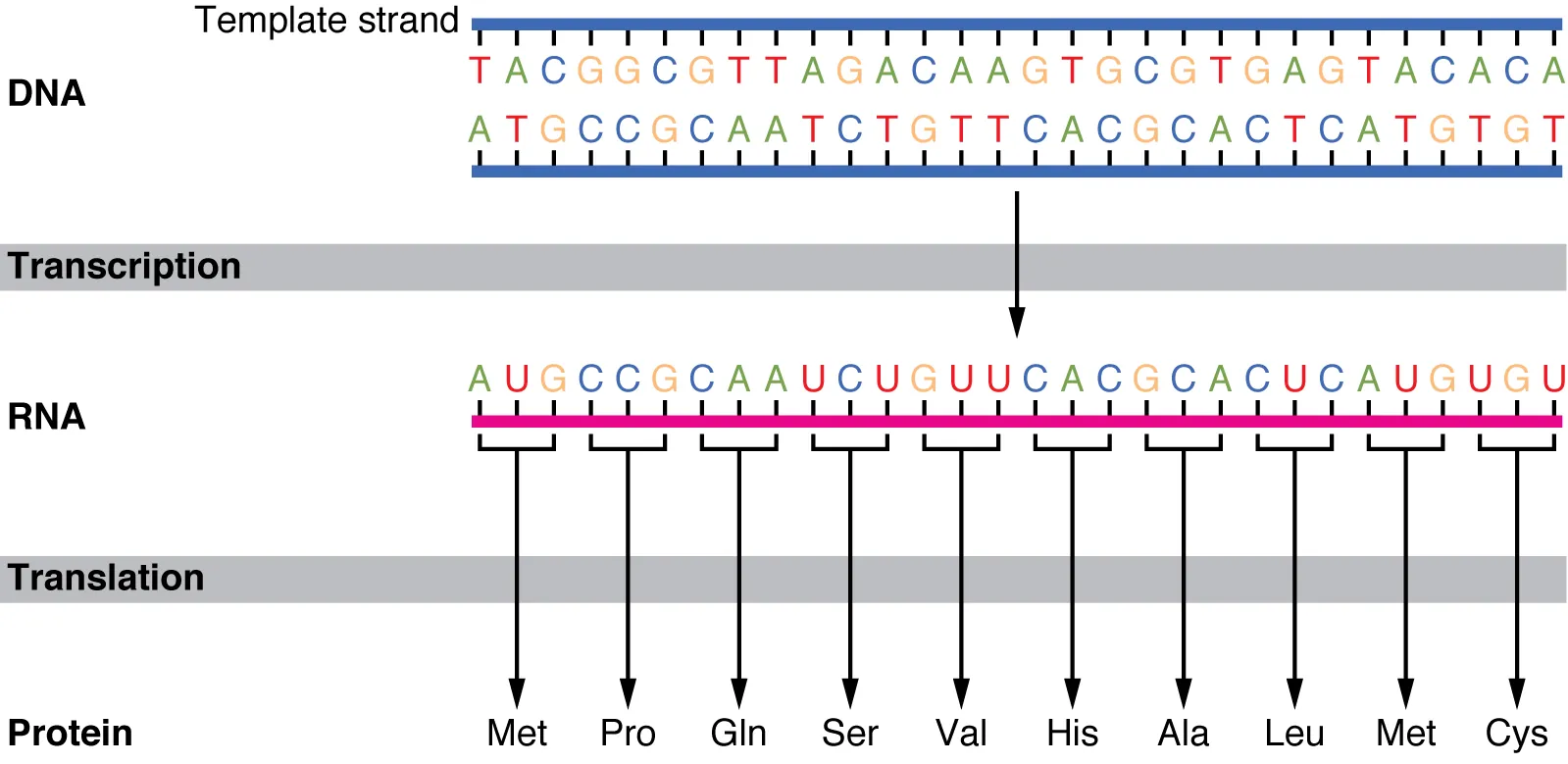
Genetic Code Features
| Feature | Description | Clinical Significance |
|---|---|---|
| Degeneracy (redundancy) | Multiple codons encode the same amino acid (64 codons for 20 AAs + stop signals) | 3rd position "wobble" allows single tRNA to recognize multiple codons; some point mutations are silent |
| Unambiguous | Each codon specifies only one amino acid | No ambiguity in translation |
| Universal | Nearly all organisms use the same code | Exceptions: mitochondria, some protists; allows recombinant DNA technology |
| Commaless & non-overlapping | Read in continuous triplets without gaps or overlaps | Frameshift mutations (insertions/deletions not multiple of 3) are devastating |
| Start codon | AUG (methionine in eukaryotes, fMet in prokaryotes) | First AUG in mRNA sets reading frame |
| Stop codons | UAA, UAG, UGA | UGA also encodes selenocysteine with SECIS element |
Types of Mutations
| Mutation Type | Description | Example |
|---|---|---|
| Silent | Nucleotide change, same amino acid (3rd position wobble) | No phenotypic effect |
| Missense | Nucleotide change, different amino acid | Sickle cell (GAG → GTG; Glu → Val) |
| Nonsense | Nucleotide change creates premature stop codon | Truncated protein; often β-thalassemia |
| Frameshift | Insertion or deletion (not multiple of 3) | Tay-Sachs (4-bp insertion in hexosaminidase A) |
| Splice site | Mutation at intron-exon junction | Some β-thalassemia mutations |
| Trinucleotide repeat expansion | Repeat sequence expands beyond threshold | Huntington (CAG), Fragile X (CGG), Myotonic dystrophy (CTG), Friedreich ataxia (GAA) |
Cell Signaling Pathways
| Pathway | Receptor Type | Key Mediators | Clinical Example |
|---|---|---|---|
| cAMP (Gs) | GPCR | Gs → adenylyl cyclase → cAMP → PKA | Cholera toxin: constitutive Gs activation → ↑cAMP → secretory diarrhea |
| cAMP (Gi) | GPCR | Gi inhibits adenylyl cyclase → ↓cAMP | Pertussis toxin: inactivates Gi → ↑cAMP → whooping cough symptoms |
| IP3/DAG (Gq) | GPCR | Gq → PLC → IP3 (↑Ca2+) + DAG (→PKC) | α1-adrenergic receptors, H1 histamine, vasopressin V1 |
| Receptor tyrosine kinase | RTK | Ras → Raf → MEK → ERK (MAP kinase cascade) | Insulin receptor, EGF receptor; oncogenes (RAS mutations in cancers) |
| JAK-STAT | Cytokine receptor | JAK phosphorylates STATs → dimerize → nuclear translocation | Erythropoietin, growth hormone, interferons; JAK2 mutation in polycythemia vera |
| Steroid receptor | Intracellular/nuclear | Ligand-receptor complex acts as transcription factor | Cortisol, estrogen, testosterone, thyroid hormone, vitamin D |
26 Clinical Correlates & Metabolic Integration
Metabolic pathways do not operate in isolation; they are interconnected, hormonally regulated, and tissue-specific. Understanding the fed vs. fasted metabolic states and the integration of major pathways is essential for clinical reasoning.
Fed vs. Fasted State
| Parameter | Fed State (Insulin High) | Fasting State (Glucagon High) |
|---|---|---|
| Liver | Glycogenesis, lipogenesis, protein synthesis | Glycogenolysis, gluconeogenesis, ketogenesis, β-oxidation |
| Muscle | Glucose uptake (GLUT4), glycogenesis, protein synthesis | Proteolysis (Ala → liver for gluconeogenesis), fatty acid oxidation |
| Adipose | Lipogenesis, glucose uptake (GLUT4), LPL activity ↑ | Lipolysis (hormone-sensitive lipase activated by glucagon/epi) |
| Brain | Glucose as primary fuel (GLUT1, insulin-independent) | Glucose initially; ketone bodies during prolonged fasting (>2–3 days) |
| RBCs | Glycolysis only (no mitochondria) | Glycolysis only; glucose is essential fuel |
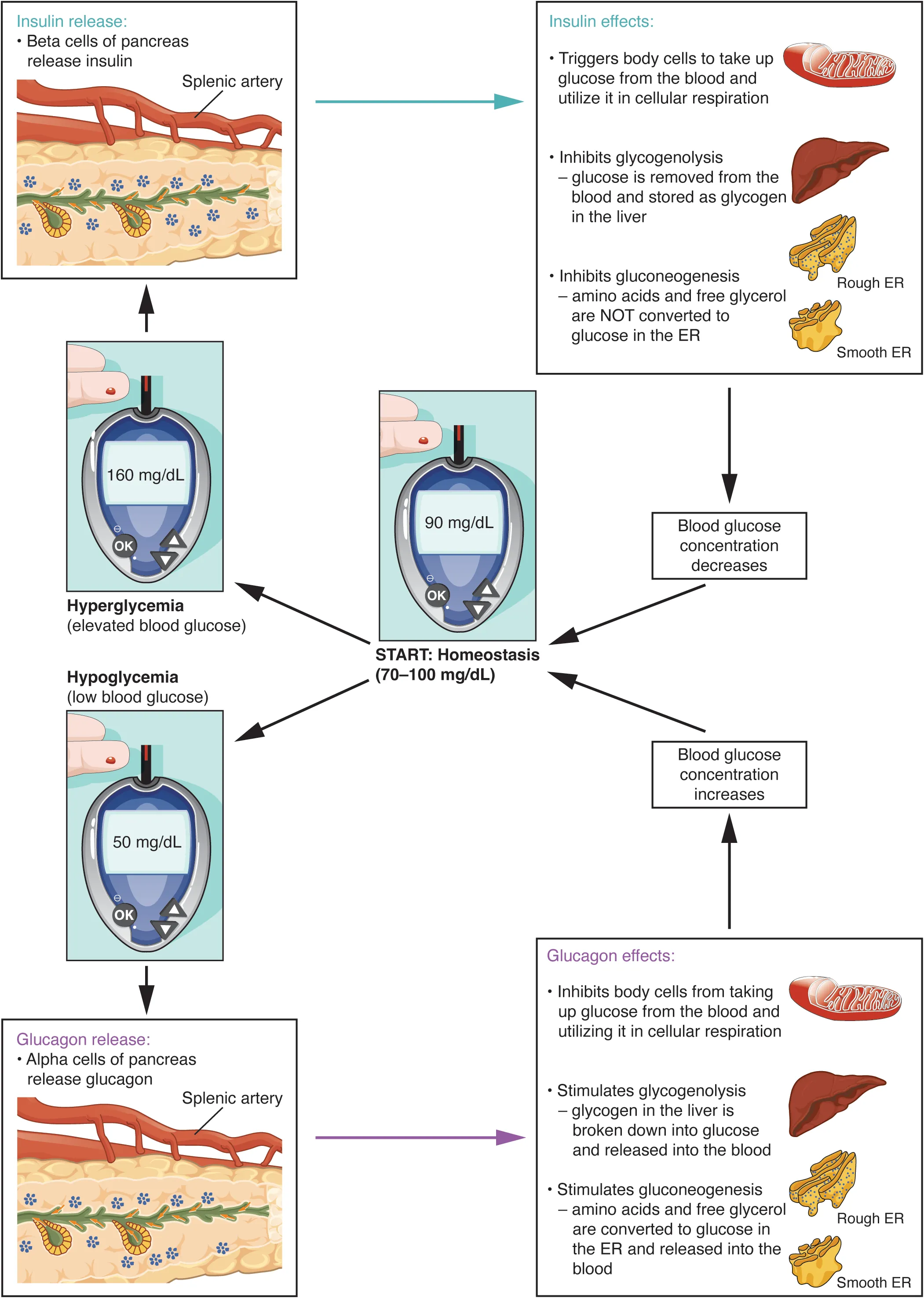
Rate-Limiting Enzymes of Major Pathways
| Pathway | Rate-Limiting Enzyme | Key Regulators |
|---|---|---|
| Glycolysis | PFK-1 | AMP, F2,6BP (↑); ATP, citrate (↓) |
| Gluconeogenesis | Fructose-1,6-bisphosphatase | Citrate, ATP (↑); AMP, F2,6BP (↓) |
| TCA cycle | Isocitrate dehydrogenase | ADP (↑); ATP, NADH (↓) |
| Glycogenesis | Glycogen synthase | Insulin, G6P (↑); glucagon, epi (↓) |
| Glycogenolysis | Glycogen phosphorylase | Glucagon, epi, AMP (↑); insulin, G6P, ATP (↓) |
| FA synthesis | Acetyl-CoA carboxylase (ACC) | Insulin, citrate (↑); glucagon, palmitoyl-CoA (↓) |
| β-Oxidation | CPT-I | Glucagon (↑); malonyl-CoA (↓) |
| Ketogenesis | HMG-CoA synthase | ↑ in fasting/starvation/DKA |
| Cholesterol synthesis | HMG-CoA reductase | Insulin (↑); glucagon, cholesterol (↓); target of statins |
| Urea cycle | CPS I | N-acetylglutamate (↑) |
| PPP | G6PD | NADP+ (↑); NADPH (↓) |
| Pyrimidine synthesis | CPS II | ATP, PRPP (↑); UTP (↓) |
| Purine synthesis | Glutamine-PRPP amidotransferase | PRPP (↑); AMP, GMP, IMP (↓) |
| Heme synthesis | ALA synthase | Heme (↓, feedback); B6 (cofactor) |
Metabolic Fuel Sources by Duration of Fasting
0–4 hours: dietary glucose, hepatic glycogenolysis. 4–16 hours: hepatic glycogenolysis, gluconeogenesis begins. 16–48 hours: hepatic glycogen depleted; gluconeogenesis (from amino acids, glycerol, lactate) is primary glucose source; fatty acid oxidation increases. >2–3 days: ketogenesis accelerates; brain switches to ketone bodies (sparing glucose and muscle protein). Weeks of starvation: ketones supply ~75% of brain fuel; muscle proteolysis slows to preserve lean mass.
Tissue-Specific Metabolism
| Tissue | Preferred Fuel(s) | Key Metabolic Features |
|---|---|---|
| Brain | Glucose (normally); ketones (fasting) | Cannot use fatty acids (don't cross BBB); GLUT1/3 (insulin-independent); high O2 demand |
| RBCs | Glucose (exclusively) | No mitochondria → only glycolysis; no TCA, no ETC, no β-oxidation; produce 2,3-BPG via Rapoport-Luebering shunt |
| Liver | Amino acids, fatty acids, glucose | Metabolic hub: gluconeogenesis, ketogenesis, ureagenesis, bile acid synthesis, VLDL production. Glucokinase (not hexokinase), has glucose-6-phosphatase |
| Skeletal muscle | Fatty acids (rest); glucose (exercise) | GLUT4 (insulin-dependent); glycogen for local use only (no G6Pase); produces lactate (anaerobic) and alanine (Cori/alanine cycles) |
| Cardiac muscle | Fatty acids (>70%), ketones, lactate, glucose | Highly aerobic (abundant mitochondria); can use lactate as fuel (LDH-1 predominant); most versatile fuel use |
| Adipose tissue | Fatty acids | GLUT4; stores TGs; hormone-sensitive lipase releases FFAs in fasting; lipoprotein lipase on capillaries clears circulating TGs |
| Kidney | Fatty acids, glutamine | Gluconeogenesis (especially in prolonged fasting); NH4+ excretion from glutamine for acid-base balance |
Ethanol is metabolized by alcohol dehydrogenase (ADH) and then aldehyde dehydrogenase (ALDH), generating excess NADH. The high NADH/NAD+ ratio shifts equilibria: (1) Pyruvate → lactate (lactic acidosis). (2) OAA → malate (inhibits gluconeogenesis → fasting hypoglycemia). (3) DHAP → glycerol-3-phosphate (promotes TG synthesis → fatty liver). (4) Inhibits β-oxidation. Clinical: alcoholic fatty liver, hypoglycemia, lactic acidosis, hyperuricemia (lactate competes with urate for renal excretion).
Insulin & Glucagon Signaling Integration
| Hormone | Signal | Metabolic Effects |
|---|---|---|
| Insulin | Tyrosine kinase receptor → IRS-1 → PI3K → Akt (PKB) → GLUT4 translocation; also RAS-MAPK for growth | ↑ Glycolysis, glycogenesis, lipogenesis, protein synthesis, K+ uptake. ↓ Gluconeogenesis, glycogenolysis, lipolysis, ketogenesis. |
| Glucagon | GPCR → Gs → adenylyl cyclase → cAMP → PKA | ↑ Glycogenolysis, gluconeogenesis, lipolysis, β-oxidation, ketogenesis. ↓ Glycolysis, glycogenesis, lipogenesis. |
| Epinephrine | Same as glucagon in liver; β2 (muscle/liver) and α1 (IP3/Ca2+) | ↑ Glycogenolysis (muscle & liver), lipolysis, gluconeogenesis. Muscle: ↑ glycolysis for immediate energy. |
| Cortisol | Nuclear receptor → gene transcription | ↑ Gluconeogenesis (PEPCK induction), proteolysis, lipolysis. Permissive for glucagon/epi effects. Chronic: hyperglycemia, central obesity, immunosuppression. |
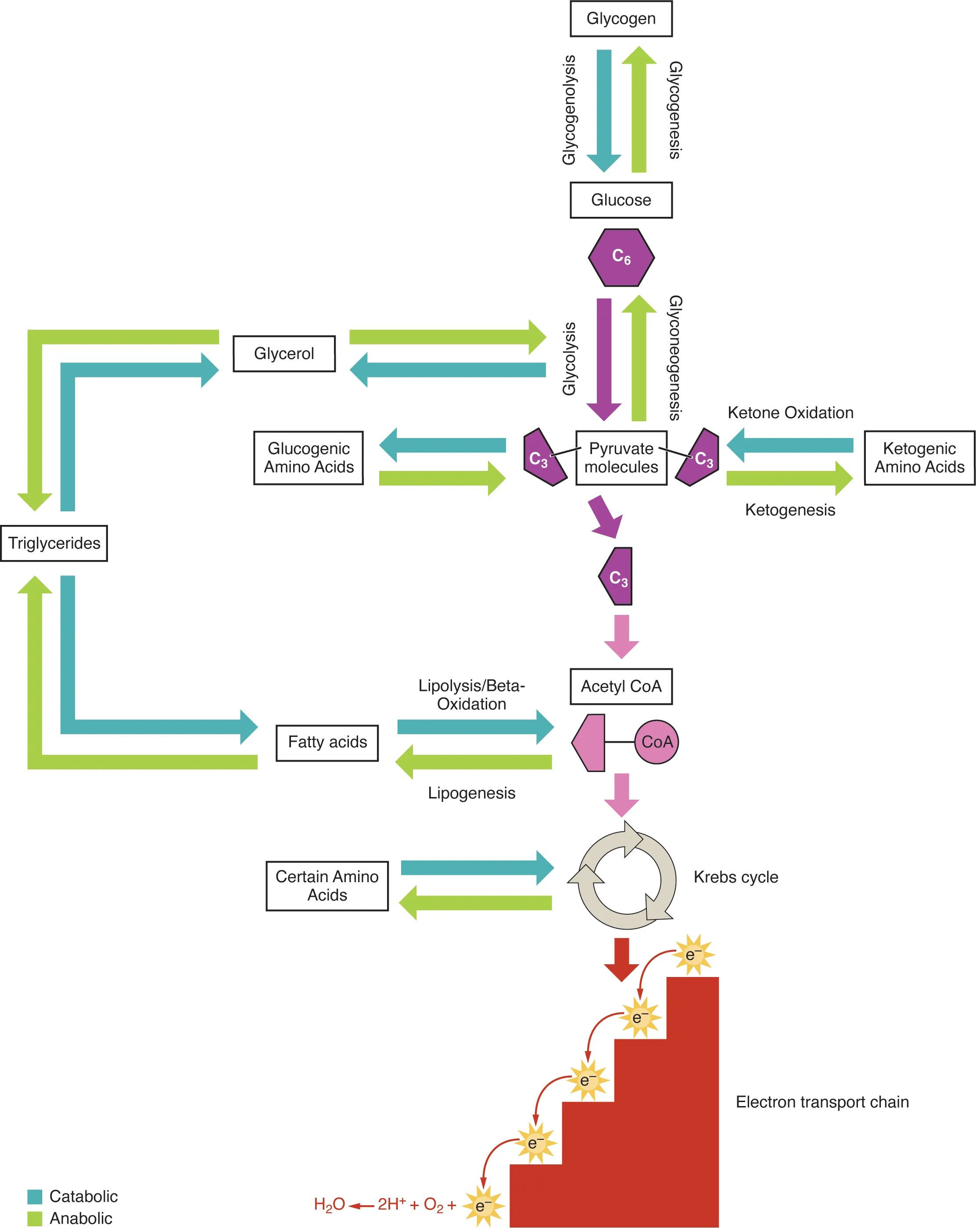
GLUT Transporters
| Transporter | Tissue(s) | Key Features |
|---|---|---|
| GLUT1 | RBCs, brain, cornea, placenta | Basal glucose uptake; insulin-independent; constitutively active |
| GLUT2 | Liver, β-islet cells, kidney, small intestine | Bidirectional, high capacity, low affinity; "glucose sensor" in β-cells |
| GLUT3 | Neurons | Low Km (high affinity); ensures brain gets glucose even at low levels |
| GLUT4 | Adipose, skeletal muscle | Insulin-dependent: insulin triggers GLUT4 vesicle fusion with membrane. Defective in type 2 DM (insulin resistance). |
| GLUT5 | Small intestine (enterocytes), spermatozoa | Fructose transporter (not glucose) |
| SGLT1 | Small intestine | Na+/glucose cotransporter (active transport); also galactose |
| SGLT2 | Proximal tubule (kidney) | Reabsorbs ~90% filtered glucose; target of SGLT2 inhibitors (empagliflozin, dapagliflozin) |
Diabetes Mellitus: Biochemical Basis
| Feature | Type 1 DM | Type 2 DM |
|---|---|---|
| Pathogenesis | Autoimmune destruction of β-cells (anti-GAD, anti-islet cell, anti-insulin antibodies) | Insulin resistance → relative insulin deficiency; β-cell dysfunction over time |
| Onset | Usually childhood/adolescence (can occur at any age) | Usually adults (increasingly in obese adolescents) |
| Insulin level | Absent / very low | Normal or elevated early; decreased late |
| C-peptide | Low / absent (no endogenous insulin) | Normal or elevated |
| DKA risk | High (absolute insulin deficiency) | Rare (some insulin present); hyperosmolar hyperglycemic state (HHS) more typical |
| Metabolic state | Catabolic: ↑lipolysis, ↑β-oxidation, ↑ketogenesis, ↑proteolysis, ↑gluconeogenesis | Anabolic: ↑TG storage, weight gain; metabolic syndrome |
| Treatment | Insulin (mandatory) | Lifestyle + metformin (first-line); may need insulin eventually |
Non-Enzymatic Glycation
In hyperglycemia, glucose non-enzymatically attaches to proteins (Maillard reaction), forming advanced glycation end products (AGEs). AGEs cross-link collagen in vessel walls, trap LDL (accelerating atherosclerosis), activate RAGE receptors (inducing inflammation and oxidative stress), and thicken basement membranes. This underlies diabetic microvascular complications: retinopathy, nephropathy, and neuropathy. HbA1c is a glycated hemoglobin that reflects average blood glucose over 2–3 months (RBC lifespan ~120 days). Target HbA1c <7% in most diabetic patients.
Four Pathways of Diabetic Damage
Chronic hyperglycemia damages tissues through four major biochemical mechanisms:
| Pathway | Mechanism | Consequence |
|---|---|---|
| Polyol (sorbitol) pathway | Aldose reductase converts glucose → sorbitol (uses NADPH); sorbitol dehydrogenase converts sorbitol → fructose | Osmotic cell damage (cataracts, neuropathy); NADPH depletion → oxidative stress |
| Advanced glycation end products | Non-enzymatic glycation of proteins and lipids | Vascular stiffening, inflammation, atherosclerosis, basement membrane thickening |
| Protein kinase C (PKC) activation | Hyperglycemia → ↑DAG → PKC activation | Altered vascular permeability, angiogenesis, endothelial dysfunction |
| Hexosamine pathway | Excess fructose-6-phosphate → glucosamine-6-phosphate → UDP-GlcNAc → O-GlcNAcylation of proteins | Altered gene expression, TGF-β upregulation, vascular smooth muscle proliferation |
27 High-Yield Review & Reference Tables
This section consolidates the most frequently tested biochemistry concepts for rapid board review.
Inborn Errors Quick-Reference
| Disease | Enzyme Defect | "Buzzword" Finding |
|---|---|---|
| PKU | Phenylalanine hydroxylase | Musty body odor, mousey urine |
| Maple syrup urine disease | Branched-chain α-ketoacid DH | Sweet/maple urine odor |
| Alkaptonuria | Homogentisic acid oxidase | Dark urine, ochronosis |
| Homocystinuria | Cystathionine β-synthase | Downward lens subluxation, thrombosis |
| Cystinuria | Dibasic AA transporter | Hexagonal urinary crystals |
| Von Gierke (GSD I) | Glucose-6-phosphatase | Severe fasting hypoglycemia, hepatomegaly |
| Pompe (GSD II) | Acid maltase | Cardiomegaly, hypotonia |
| McArdle (GSD V) | Muscle glycogen phosphorylase | No lactate rise on ischemic forearm test |
| Tay-Sachs | Hexosaminidase A | Cherry-red macula, no HSM |
| Gaucher | Glucocerebrosidase | "Crumpled tissue paper" macrophages |
| Niemann-Pick A | Sphingomyelinase | Cherry-red macula + HSM |
| Fabry | α-Galactosidase A | Angiokeratomas, burning extremity pain |
| Hurler (MPS I) | α-L-iduronidase | Corneal clouding, gargoylism |
| Hunter (MPS II) | Iduronate-2-sulfatase | NO corneal clouding, X-linked |
| Lesch-Nyhan | HGPRT | Self-mutilation, hyperuricemia |
| G6PD deficiency | Glucose-6-phosphate DH | Heinz bodies, bite cells |
| Hereditary fructose intolerance | Aldolase B | Hypoglycemia after fructose ingestion |
| Classic galactosemia | Gal-1-P uridylyltransferase | Cataracts, E. coli sepsis in neonate |
Vitamin Deficiency Quick-Reference
| Vitamin | Deficiency Buzzword |
|---|---|
| A | Night blindness, Bitot spots, xerophthalmia |
| B1 (thiamine) | Beriberi, Wernicke-Korsakoff |
| B2 (riboflavin) | Cheilosis, corneal vascularization |
| B3 (niacin) | Pellagra (3 D's: Diarrhea, Dermatitis, Dementia) |
| B5 (pantothenate) | Burning feet, dermatitis (rare) |
| B6 (pyridoxine) | Sideroblastic anemia, seizures (INH use) |
| B7 (biotin) | Dermatitis, alopecia (raw eggs/avidin) |
| B9 (folate) | Megaloblastic anemia, neural tube defects |
| B12 (cobalamin) | Megaloblastic anemia + subacute combined degeneration, ↑MMA |
| C (ascorbic acid) | Scurvy: bleeding gums, petechiae, corkscrew hairs |
| D | Rickets (children), osteomalacia (adults) |
| E | Hemolytic anemia, spinocerebellar degeneration |
| K | Bleeding diathesis (↑PT), hemorrhagic disease of newborn |
Key Cofactor Associations
| Cofactor | Vitamin Source | Key Enzymes |
|---|---|---|
| TPP | B1 (thiamine) | PDH, α-KG DH, transketolase, branched-chain α-ketoacid DH |
| FAD / FMN | B2 (riboflavin) | Succinate DH (Complex II), NADH DH (Complex I), acyl-CoA DH |
| NAD+ / NADP+ | B3 (niacin) | Glycolysis, TCA cycle, ETC, PPP, gluconeogenesis |
| CoA | B5 (pantothenate) | PDH, α-KG DH, fatty acid synthesis/oxidation |
| PLP | B6 (pyridoxine) | Transaminases, ALA synthase, glycogen phosphorylase, decarboxylases |
| Biotin | B7 | Pyruvate carboxylase, ACC, propionyl-CoA carboxylase |
| THF | B9 (folate) | Thymidylate synthase, purine synthesis, methionine synthase |
| Deoxyadenosylcobalamin / Methylcobalamin | B12 | Methylmalonyl-CoA mutase, methionine synthase |
Metabolic Pathway Locations
| Pathway | Cellular Location |
|---|---|
| Glycolysis | Cytoplasm |
| Gluconeogenesis | Cytoplasm (mostly) + mitochondria (pyruvate carboxylase) + ER (G6Pase) |
| TCA cycle | Mitochondrial matrix |
| Oxidative phosphorylation | Inner mitochondrial membrane |
| Fatty acid synthesis | Cytoplasm |
| β-Oxidation | Mitochondrial matrix |
| Ketogenesis | Mitochondrial matrix (liver only) |
| Pentose phosphate pathway | Cytoplasm |
| Urea cycle | Mitochondria (CPS I, OTC) + cytoplasm (remaining steps) |
| Heme synthesis | Mitochondria (first & last steps) + cytoplasm (middle steps) |
| Cholesterol synthesis | Cytoplasm (SER) |
| Steroid synthesis | Mitochondria (side-chain cleavage) + SER (subsequent steps) |
| Protein synthesis | RER (secreted/membrane proteins) or free ribosomes (cytoplasmic/nuclear proteins) |
| Glycogen synthesis/degradation | Cytoplasm |
| Peroxisomal β-oxidation | Peroxisomes (VLCFA, branched-chain FA) |
X-Linked Metabolic Disorders
| Disorder | Defect | Key Feature |
|---|---|---|
| G6PD deficiency | Glucose-6-phosphate dehydrogenase | Hemolytic anemia with oxidant stress |
| Fabry disease | α-Galactosidase A | Angiokeratomas, renal failure, neuropathy |
| Hunter syndrome (MPS II) | Iduronate-2-sulfatase | Hurler-like but no corneal clouding |
| Lesch-Nyhan syndrome | HGPRT | Hyperuricemia, self-mutilation |
| OTC deficiency | Ornithine transcarbamylase | Hyperammonemia, elevated orotic acid |
| Menkes disease | ATP7A (copper transporter) | Kinky hair, connective tissue laxity |
| Adrenoleukodystrophy | ABCD1 (peroxisomal VLCFA transporter) | Adrenal insufficiency, CNS demyelination |
| Pyruvate dehydrogenase deficiency | PDH E1 subunit | Lactic acidosis, neurological deficits |
Inheritance Patterns in Metabolic Disease
The vast majority of inborn errors of metabolism follow autosomal recessive inheritance: both parents are carriers, and each offspring has a 25% chance of being affected. Notable X-linked exceptions are listed above. Autosomal dominant metabolic disorders are rare but include familial hypercholesterolemia (LDL receptor), acute intermittent porphyria (PBG deaminase), and some forms of hereditary spherocytosis.
Laboratory Values & Metabolic Panels
| Lab Test | Normal Range | Elevated In | Decreased In |
|---|---|---|---|
| Glucose (fasting) | 70–100 mg/dL | Diabetes, Cushing, acromegaly, pheochromocytoma | Insulinoma, adrenal insufficiency, liver failure, GSD I |
| HbA1c | <5.7% | Uncontrolled diabetes (reflects 2–3 month glucose average) | Hemolytic anemia, blood loss (falsely low) |
| Ammonia | 10–35 μmol/L | Urea cycle defects, liver failure, Reye syndrome, valproic acid | Not clinically significant |
| Uric acid | 3.5–7.2 mg/dL (M) | Gout, Lesch-Nyhan, tumor lysis syndrome, von Gierke (GSD I) | SIADH, allopurinol/febuxostat therapy |
| Lactate | 0.5–2.0 mEq/L | Tissue hypoperfusion (shock), mitochondrial disease, cyanide, CO poisoning, PDH deficiency, GSD I | Not clinically significant |
| Methylmalonic acid | <0.4 μmol/L | B12 deficiency, methylmalonyl-CoA mutase deficiency | Not clinically significant |
| Homocysteine | 5–15 μmol/L | B12 deficiency, folate deficiency, homocystinuria, B6 deficiency | Not clinically significant |
Musty/mousey odor → PKU. Sweet/maple syrup odor → MSUD. Dark urine → alkaptonuria. Cherry-red macula → Tay-Sachs, Niemann-Pick (or CRAO). Hexagonal crystals → cystinuria. Crumpled tissue paper macrophages → Gaucher. Heinz bodies & bite cells → G6PD deficiency. Gargoylism + corneal clouding → Hurler. Self-mutilation → Lesch-Nyhan. Port-wine urine → acute intermittent porphyria. Fruity breath → DKA. Corkscrew hairs → scurvy. Casal necklace → pellagra (B3 deficiency).
Tumor Metabolism
| Concept | Description | Clinical Application |
|---|---|---|
| Warburg effect | Aerobic glycolysis: cancer cells prefer glycolysis even with adequate O2 | Basis for FDG-PET imaging (increased glucose uptake) |
| Oncometabolites | IDH1/2 mutations produce 2-hydroxyglutarate (2-HG), an abnormal metabolite | 2-HG inhibits α-KG-dependent enzymes → hypermethylation → AML, glioma |
| Tumor lysis syndrome | Rapid cell death releases purines → xanthine oxidase → massive uric acid production | Hyperuricemia, hyperkalemia, hyperphosphatemia, hypocalcemia. Prevent with allopurinol or rasburicase. |
| Glutamine addiction | Many cancers are dependent on glutamine for anaplerosis and biosynthesis | Glutaminase inhibitors under investigation as anticancer agents |