Gametogenesis, fertilization, gastrulation, organogenesis, germ layer derivatives, congenital anomalies, teratology, fetal development, and every developmental mechanism, signaling pathway, and clinical malformation across the full scope of medical embryology.
01 Overview & Significance
Embryology is the study of human development from fertilization through birth. Understanding embryologic processes is essential for explaining congenital anomalies, anatomical relationships, and the rational basis for surgical repair. Approximately 3% of live-born infants have a clinically significant birth defect, and developmental errors account for a large fraction of pediatric hospitalizations and neonatal mortality.
Why This Matters
A strong foundation in embryology is essential for clinical reasoning across every specialty. Surgeons must understand normal developmental anatomy to repair congenital defects. Pediatricians and geneticists use embryologic principles to diagnose and counsel families. Obstetricians rely on knowledge of teratogens and fetal development for prenatal care. Board examinations heavily test embryologic origins and associated malformations.
Gastrulation, neurulation, organogenesis; maximum teratogen susceptibility
Fetal
Weeks 9–38
Growth, maturation, functional development; teratogens cause growth restriction or functional defects
Organizing Principles
Cranial-to-caudal — development proceeds from head to tail (neural tube closes rostrally first)
Proximal-to-distal — limbs develop from shoulder/hip outward to digits
Induction — one tissue signals another to change fate (e.g., notochord induces overlying ectoderm to form neural plate)
Apoptosis — programmed cell death sculpts structures (e.g., digit separation, lumen formation in GI tube)
Lateral folding — embryonic disc folds to form a tubular body plan; failure causes ventral wall defects
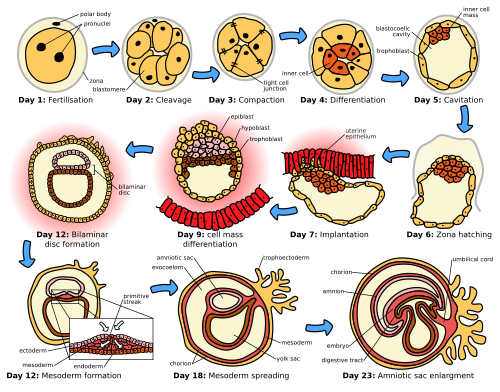
Figure 1 — Stages of Human Embryogenesis. Overview of the major developmental stages from fertilization through early organogenesis, illustrating the progressive complexity of the embryo during the pre-embryonic and embryonic periods.
All-or-nothing period: during weeks 1–2, insults either kill the embryo or cause no lasting damage because the cells are totipotent and can compensate. The most teratogen-sensitive window is weeks 3–8 (organogenesis).
02 Core Principles & Signaling
Embryologic development is orchestrated by a conserved set of signaling molecules, transcription factors, and morphogen gradients. Mutations in these pathways underlie many congenital syndromes.
Major Signaling Pathways
Pathway
Role in Development
Associated Defect
Sonic Hedgehog (SHH)
Ventral patterning of neural tube, limb bud ZPA, midline face development
Hox genes specify anterior-posterior positional identity along the body axis. They are arranged in clusters (A–D) and are expressed in a colinear fashion: genes at the 3′ end of the cluster are expressed earlier and more anteriorly than those at the 5′ end. Mutations cause homeotic transformations (e.g., a vertebral segment adopts the identity of a more anterior or posterior one). The HOXA13 mutation causes hand-foot-genital syndrome.
Key Transcription Factors
PAX genes: PAX2 (kidney), PAX3 (Waardenburg syndrome), PAX6 (aniridia, eye development), PAX9 (teeth). SOX9: chondrogenesis, sex determination. SRY: testis-determining factor on Y chromosome. TBX5: upper limb and heart (Holt-Oram syndrome). TBX4: lower limb development.
Morphogen Gradients & Axes
Axis
Key Molecules
Mechanism
Dorsal-ventral (neural tube)
SHH (ventral, from notochord/floor plate); BMP/Wnt (dorsal, from roof plate)
Concentration gradient specifies motor neurons ventrally, sensory interneurons dorsally
Maintains proliferation of progress zone mesenchyme; AER removal → limb truncation
Dorsal-ventral (limb)
Wnt7a (dorsal ectoderm)
Specifies dorsal limb structures; loss → ventral duplication
03 Gametogenesis
Gametogenesis is the formation of haploid gametes (sperm and oocytes) from diploid germ cells through meiosis. Errors during meiosis are the leading cause of chromosomal aneuploidies such as trisomy 21 (Down syndrome).
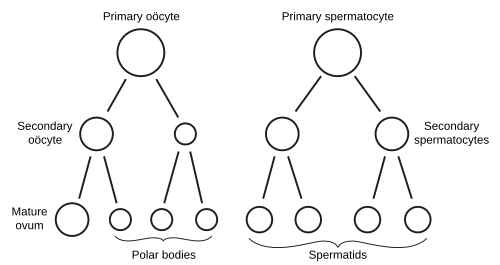
Figure 2 — Gametogenesis: Oogenesis and Spermatogenesis. Comparison of the two gametogenesis pathways showing the progression from diploid germ cells through meiotic divisions to haploid gametes. Note the unequal cytoplasmic division in oogenesis producing one ovum and polar bodies, versus four equal spermatozoa in spermatogenesis.
Oogenesis
Oogonia undergo mitosis during fetal life; all primary oocytes are formed by month 5 of fetal development (~7 million, declining to ~2 million at birth and ~400,000 at puberty)
Primary oocytes arrest in prophase I (dictyotene) until ovulation — this arrest can last up to 50 years
At ovulation, meiosis I completes → secondary oocyte + first polar body
Meiosis II begins but arrests at metaphase II until fertilization
Fertilization triggers completion of meiosis II → mature ovum + second polar body
Increasing maternal age increases risk of nondisjunction (especially meiosis I errors), explaining the rising incidence of trisomies with advanced maternal age
Duration: ~64 days from spermatogonium to mature sperm
Spermiogenesis: final maturation step — Golgi forms acrosome, centriole forms flagellum, mitochondria form midpiece, excess cytoplasm shed as residual body
Sertoli cells provide nutritional support and form the blood-testis barrier; Leydig cells produce testosterone
Meiosis vs. Mitosis — Clinical Significance
Meiosis I separates homologous chromosomes (reductional division); nondisjunction here produces two abnormal gametes (both aneuploid). Meiosis II separates sister chromatids (equational, similar to mitosis); nondisjunction here produces one normal and one abnormal gamete pair. Most trisomy 21 cases result from maternal meiosis I nondisjunction. Crossing over during prophase I generates genetic diversity and is essential for proper chromosome segregation.
Feature
Oogenesis
Spermatogenesis
Onset
Fetal life (mitosis); puberty (meiosis resumes)
Puberty
Output per meiosis
1 ovum + 3 polar bodies
4 spermatozoa
Duration
Decades (arrested in prophase I)
~64 days per cycle
Arrest points
Prophase I (fetal → ovulation); Metaphase II (until fertilization)
None (continuous)
Cytoplasm
Unequal division — ovum gets most
Equal division
Age-related errors
Increasing nondisjunction with age
Point mutations accumulate with paternal age
Chromosomal Abnormalities from Meiotic Errors
Abnormality
Mechanism
Clinical Features
Trisomy 21 (Down syndrome)
Meiosis I nondisjunction (~95%); Robertsonian translocation (~4%); mosaicism (~1%)
Severe intellectual disability, rocker-bottom feet, clenched fists (overlapping fingers), micrognathia, cardiac defects; death usually by age 1
Trisomy 13 (Patau syndrome)
Nondisjunction or Robertsonian translocation
Holoprosencephaly, cleft lip/palate, polydactyly, microphthalmia, cardiac defects; death usually by age 1
Turner syndrome (45,X)
Loss of one X chromosome; most common cause of 1st trimester miscarriage
Short stature, webbed neck, shield chest, coarctation of aorta, bicuspid aortic valve, streak gonads, lymphedema at birth, horseshoe kidney
Klinefelter syndrome (47,XXY)
Nondisjunction (maternal or paternal meiosis I)
Tall stature, gynecomastia, small testes, infertility, ↓testosterone, ↑FSH/LH
Robertsonian translocation involves the fusion of two acrocentric chromosomes (13, 14, 15, 21, 22) at their centromeres. A balanced Robertsonian translocation carrier (e.g., 14;21) has 45 chromosomes and is phenotypically normal but has a recurrence risk for trisomy 21 offspring. If the mother is the carrier, the recurrence risk is ~10–15%; if the father, ~1–2%.
Hydatidiform Moles
Type
Karyotype
Mechanism
Features
Complete mole
46,XX (most common) or 46,XY
Empty egg fertilized by one sperm that duplicates (or two sperm); entirely paternal DNA
No fetal tissue; diffuse trophoblastic proliferation ("bunch of grapes"); very high hCG; 15–20% risk of choriocarcinoma
Partial mole
69,XXY (triploid)
Normal egg fertilized by two sperm
Fetal parts present; focal trophoblastic proliferation; less elevated hCG; low risk of malignancy
04 Fertilization & Cleavage
Fertilization normally occurs in the ampulla of the uterine tube within 24 hours of ovulation. The process restores the diploid chromosome number, determines genetic sex, and initiates cleavage.
Steps of Fertilization
Capacitation — sperm undergo functional maturation in the female reproductive tract (removal of cholesterol from membrane, increased motility)
Acrosome reaction — sperm contacts zona pellucida; acrosomal enzymes (hyaluronidase, acrosin) digest the zona
Zona pellucida binding — ZP3 glycoprotein binds sperm, triggering the acrosome reaction
Sperm-oocyte fusion — sperm membrane fuses with oocyte membrane
Completion of meiosis II — second polar body extruded; male and female pronuclei form and fuse (syngamy)
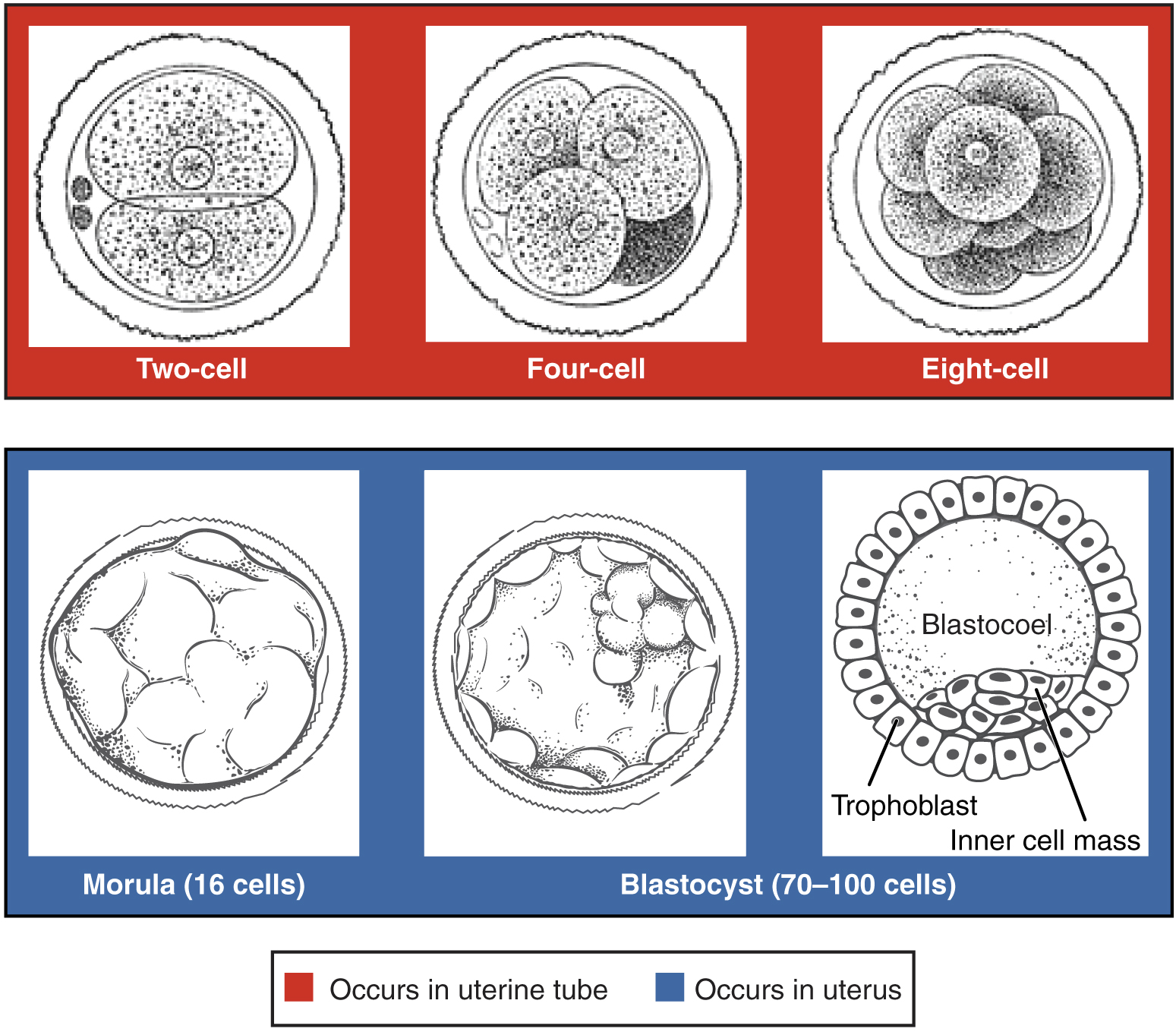
Figure 3 — Pre-Embryonic Cleavage Stages. Serial mitotic divisions of the zygote produce progressively smaller blastomeres. By day 3, a 16-cell morula forms; by day 4–5, a fluid-filled blastocoel develops within the structure, creating the blastocyst.
Cleavage & Morula Formation
After fertilization, the zygote undergoes rapid mitotic divisions (cleavage) without overall growth. Cell number increases while cell size decreases. By day 3, a 16-cell solid ball called the morula enters the uterine cavity. The morula compacts, and by day 4–5 a fluid-filled cavity (blastocoel) forms, creating the blastocyst.
Blastocyst
Structure
Fate
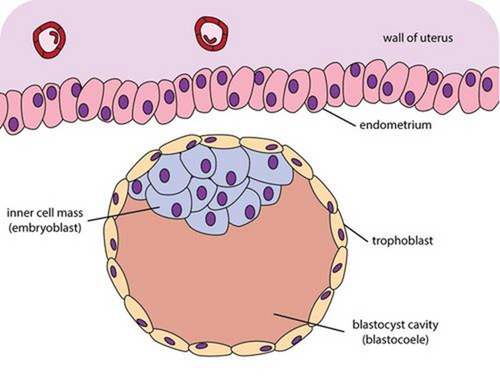
Inner cell mass (embryoblast)
Embryo proper, amnion, yolk sac, allantois
Outer cell mass (trophoblast)
Placenta (cytotrophoblast + syncytiotrophoblast)
Blastocoel
Eventually obliterated as structures expand
Figure 4 — Blastocyst Structure. The blastocyst consists of an outer trophoblast layer (which forms the placenta), an inner cell mass or embryoblast (which gives rise to the embryo proper), and a fluid-filled blastocoel cavity.
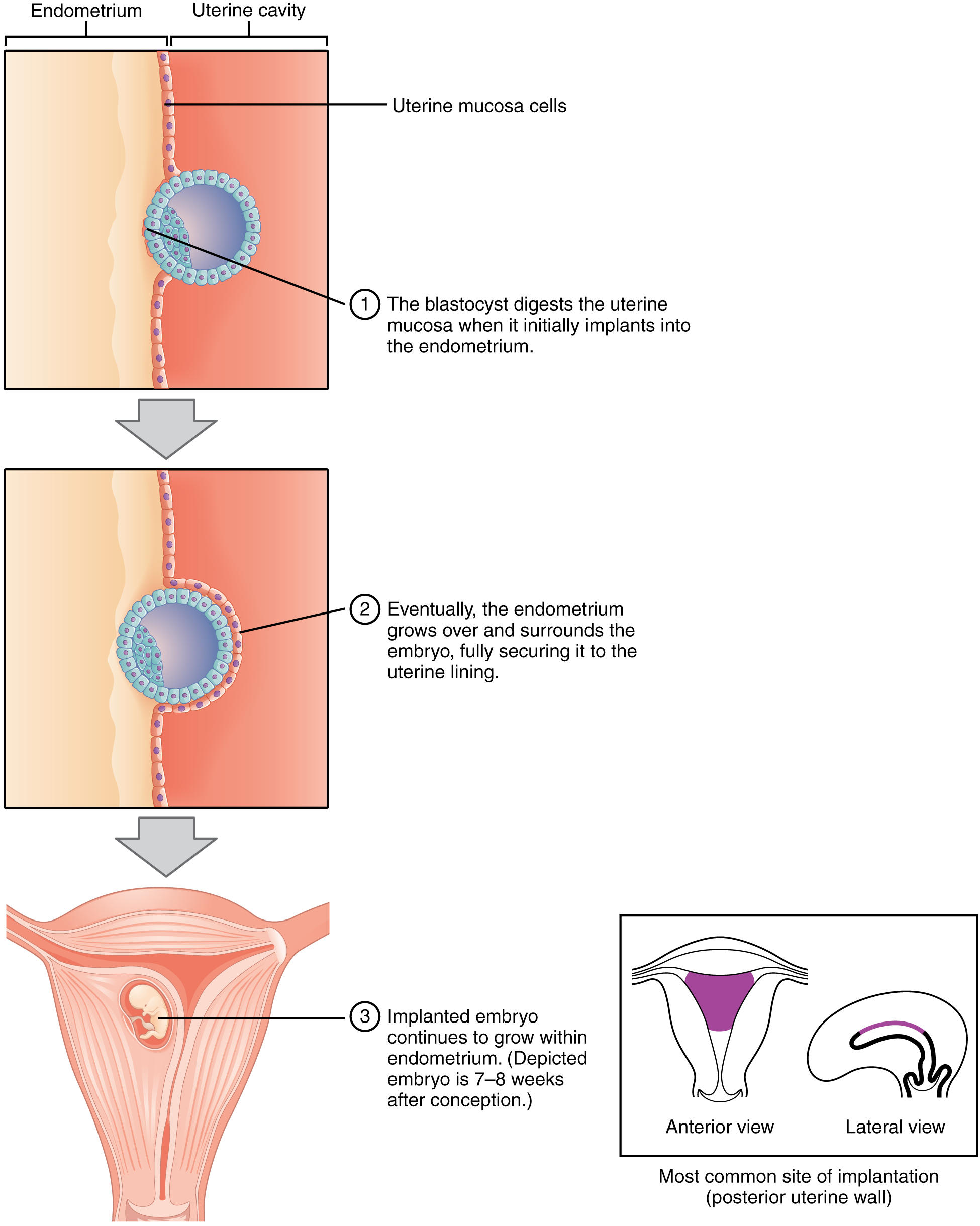
Ectopic pregnancy occurs when implantation happens outside the uterine cavity — most commonly in the ampulla of the uterine tube (~95% tubal). Risk factors include PID, prior ectopic, and tubal surgery. Ruptured ectopic is a surgical emergency causing hemoperitoneum.
05 Implantation & Bilaminar Disc
Implantation begins around day 6 when the blastocyst attaches to the posterior wall of the uterus (most common site). The syncytiotrophoblast invades the endometrial stroma, and the embryo is fully embedded by day 9–10.
Week 1 Events
Day
Event
Day 0
Fertilization in ampulla of uterine tube
Day 1–3
Cleavage divisions; morula formation
Day 4–5
Blastocyst formation; enters uterine cavity; zona pellucida degenerates (hatching)
Day 6
Implantation begins; trophoblast differentiates into cyto- and syncytiotrophoblast
Figure 5 — Implantation of the Blastocyst. The blastocyst implants into the posterior uterine wall around day 6. The syncytiotrophoblast invades the endometrial stroma, and the embryo is fully embedded by day 9–10. The cytotrophoblast and syncytiotrophoblast layers are visible surrounding the inner cell mass.
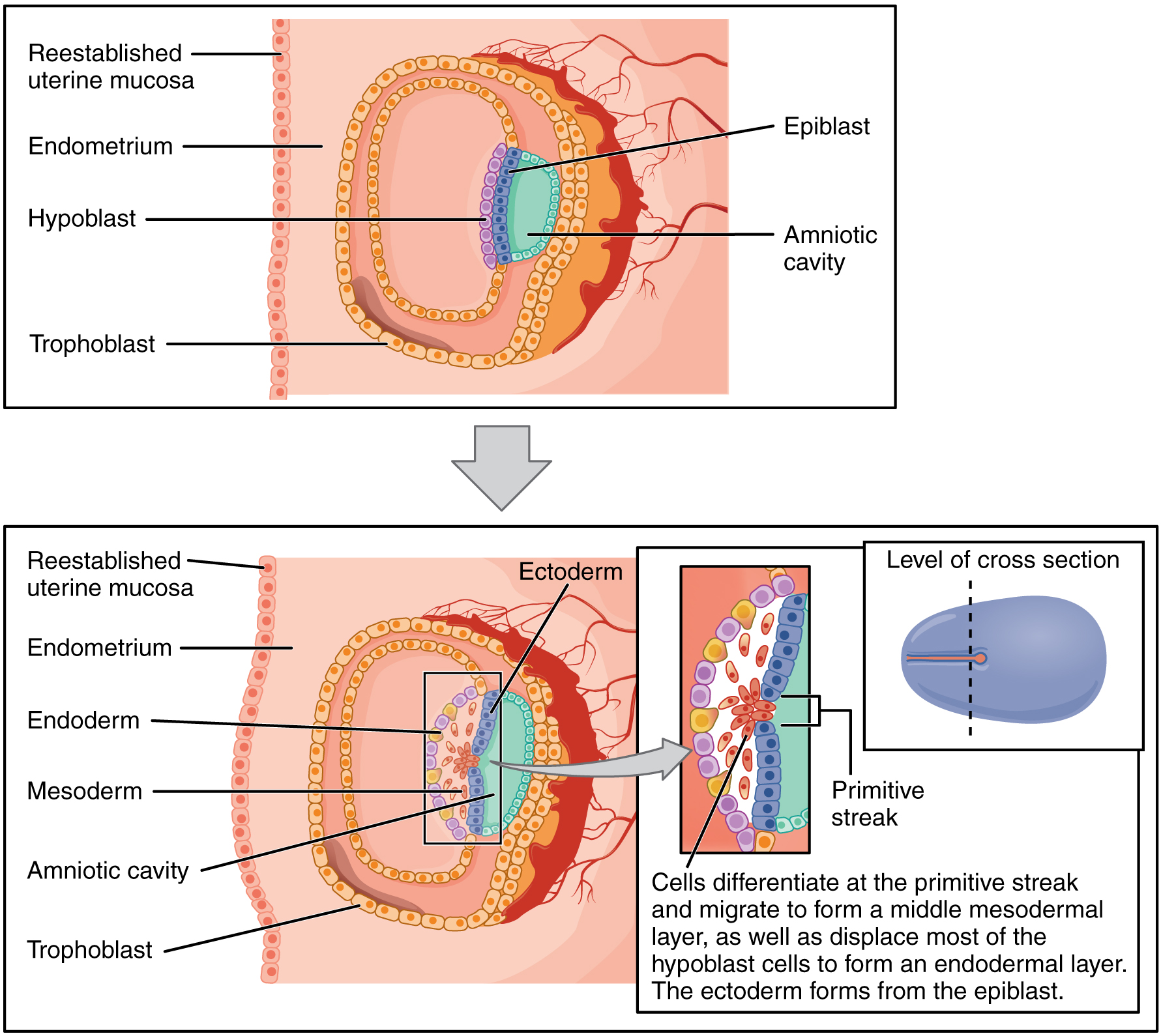
Week 2: "Rule of Twos"
Week 2 is characterized by the formation of two germ layers (epiblast + hypoblast = bilaminar disc), two cavities (amniotic cavity + yolk sac), and two trophoblast layers (cytotrophoblast + syncytiotrophoblast).
Structure
Origin
Function/Fate
Epiblast
Inner cell mass
Gives rise to all three germ layers; lines the amniotic cavity
Hypoblast
Inner cell mass
Lines the yolk sac; displaced by endoderm during gastrulation
Amniotic cavity
Within epiblast
Surrounds embryo; amniotic fluid protects and allows movement
Primary yolk sac
Hypoblast-lined
Nutrient transfer early; later forms definitive (secondary) yolk sac
Extraembryonic mesoderm
Epiblast/trophoblast
Lines chorion and amnion; forms connecting stalk (future umbilical cord)
hCG & Corpus Luteum
The syncytiotrophoblast secretes human chorionic gonadotropin (hCG) beginning at implantation. hCG maintains the corpus luteum of pregnancy, which produces progesterone to sustain the endometrium until the placenta takes over hormone production (~weeks 8–12). hCG is the basis of pregnancy tests and doubles approximately every 48 hours in early normal pregnancy. Failure to rise appropriately raises concern for ectopic pregnancy or miscarriage.
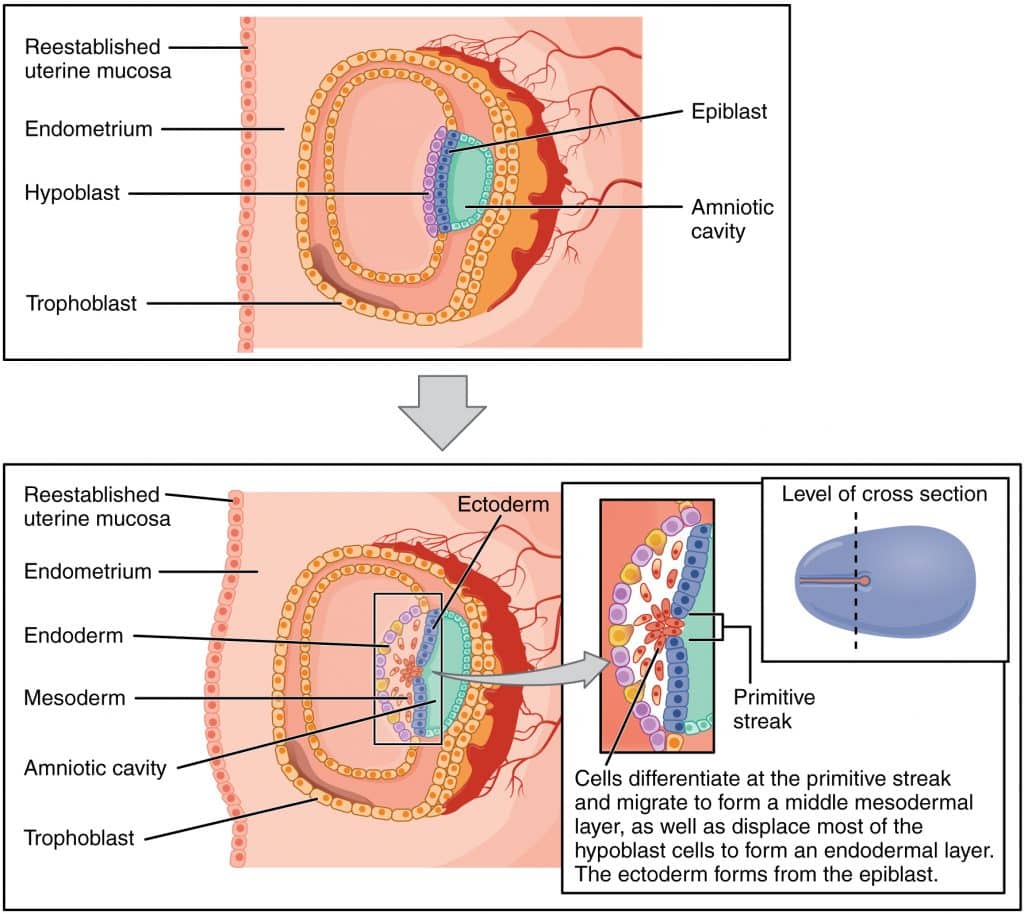
06 Gastrulation & Trilaminar Disc
Gastrulation occurs during week 3 and is the most critical event in early development. It establishes the three primary germ layers (ectoderm, mesoderm, endoderm) and the body axes (cranial-caudal, dorsal-ventral, left-right).
Figure 6 — Gastrulation: From Bilaminar to Trilaminar Disc. During week 3, epiblast cells migrate through the primitive streak to form the three primary germ layers: ectoderm, mesoderm, and endoderm. This process transforms the bilaminar disc into a trilaminar disc and establishes the body plan.
Primitive Streak & Gastrulation
The primitive streak appears on the dorsal surface of the epiblast at the caudal end
Epiblast cells migrate through the streak (ingression) — first wave displaces hypoblast to form definitive endoderm; second wave forms intraembryonic mesoderm
Remaining epiblast becomes ectoderm
The primitive node (Hensen node) at the cranial end of the streak organizes gastrulation and gives rise to the notochord
Notochord
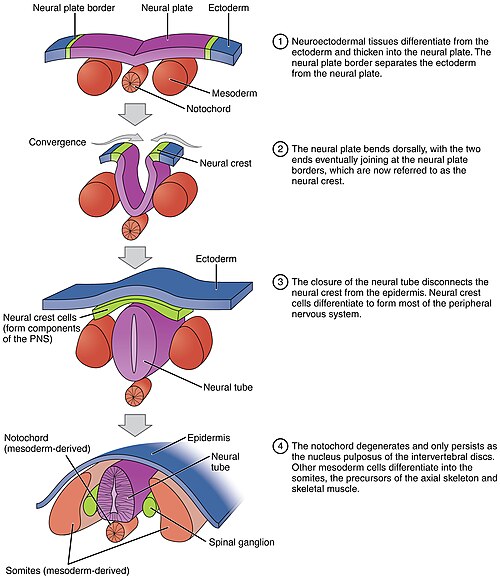
The notochord is a defining feature of all chordates. It extends from the primitive node cranially, inducing the overlying ectoderm to form the neural plate. It also signals ventral patterning of the neural tube via Sonic Hedgehog (SHH). In the adult, the notochord persists only as the nucleus pulposus of the intervertebral discs.
Week 3: "Rule of Threes"
Three germ layers form (ectoderm, mesoderm, endoderm). Three key structures arise: primitive streak, notochord, and neural plate. The allantois (endodermal outgrowth) contributes to the umbilical vessels. Gastrulation determines the body plan — disruption causes the most devastating congenital anomalies. Sacrococcygeal teratoma arises from remnants of the primitive streak that fail to regress.
Left-Right Axis Determination
Establishment of the left-right body axis occurs at the primitive node during gastrulation. Motile cilia on nodal cells generate a leftward fluid flow (nodal flow), which activates the Nodal signaling cascade on the left side of the embryo. Nodal and Lefty expression on the left side induces PITX2 transcription factor, which determines left-sided organ identity (heart looping to the right, spleen on the left, stomach on the left).
Condition
Mechanism
Features
Situs inversus totalis
Complete mirror reversal of all viscera
Usually asymptomatic; dextrocardia with reversal of all abdominal organs
Situs inversus with Kartagener syndrome
Dynein arm defect → immotile cilia (cannot generate nodal flow)
Situs inversus + bronchiectasis + chronic sinusitis (impaired mucociliary clearance); male infertility (immotile sperm)
Heterotaxy (situs ambiguus)
Partial laterality defect
Complex congenital heart disease, polysplenia or asplenia, intestinal malrotation
Kartagener syndrome (a form of primary ciliary dyskinesia) connects multiple clinical findings through a single mechanism: immotile cilia. The same dynein arm defect that prevents nodal flow (causing situs inversus) also impairs respiratory cilia (sinusitis, bronchiectasis) and sperm motility (infertility). ~50% of patients with primary ciliary dyskinesia have situs inversus.
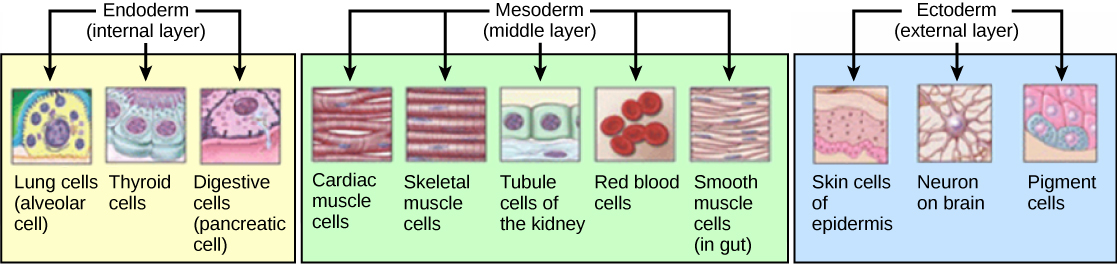
Figure 7 — Germ Layer Derivatives Overview. The three primary germ layers give rise to all tissues of the body: ectoderm (nervous system, epidermis), mesoderm (muscle, bone, cardiovascular, urogenital), and endoderm (GI epithelium, respiratory epithelium, glandular organs).
The primitive streak normally regresses by week 4. Persistence of primitive streak cells can give rise to a sacrococcygeal teratoma, the most common tumor of the newborn. It contains tissues from all three germ layers.
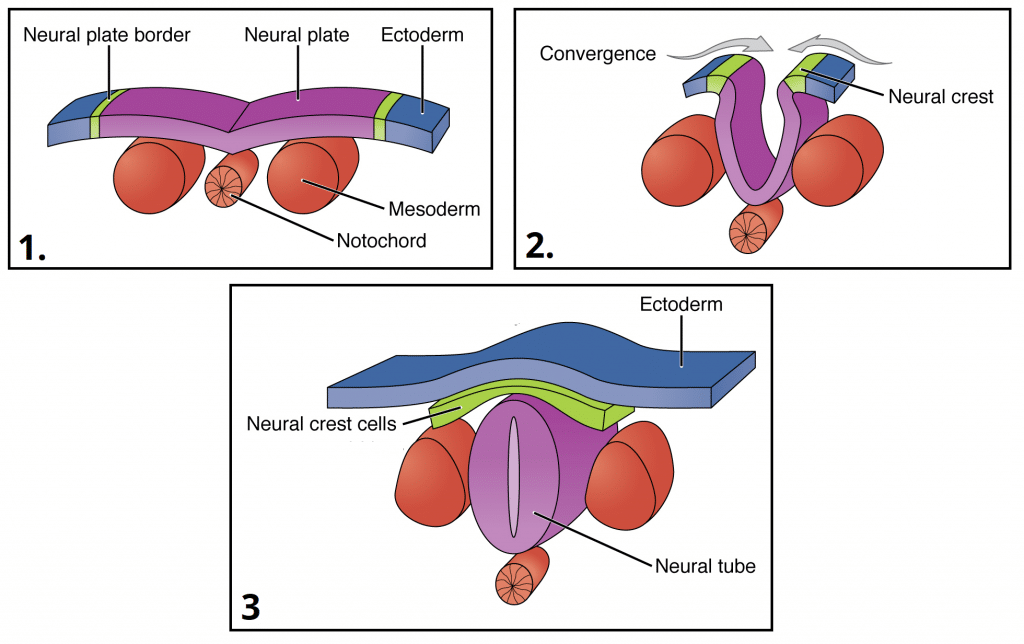
07 Neurulation & Neural Tube
Neurulation is the process by which the neural plate folds to form the neural tube, the precursor to the entire CNS. It begins during week 3 and the neural tube closes by the end of week 4.
Figure 8 — Neurulation Process. The neural plate folds to form the neural groove, then the neural folds fuse at the midline to create the neural tube. Neural crest cells delaminate from the junction of the neural tube and surface ectoderm and migrate throughout the embryo.
Steps of Neurulation
Neural plate forms from ectoderm induced by the underlying notochord
Neural plate edges elevate to form neural folds; the center depresses to form the neural groove
Neural folds fuse at the midline forming the neural tube (closure begins at the cervical region and extends cranially and caudally)
Cranial neuropore closes by day 25; caudal neuropore closes by day 28
Neural crest cells delaminate from the junction of neural tube and surface ectoderm
Figure 9 — Neural Tube Closure. Closure of the neural tube begins in the cervical region and extends bidirectionally. The cranial neuropore closes by day 25 and the caudal neuropore by day 28. Failure of closure produces neural tube defects.
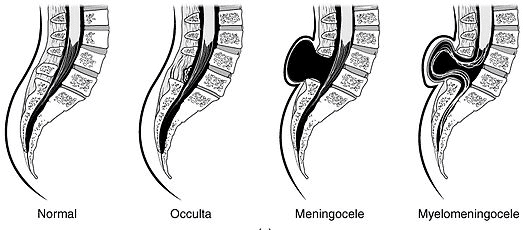
Neural Tube Defects (NTDs)
Defect
Neuropore
Description
Marker
Anencephaly
Cranial (anterior)
Failure of cranial neuropore to close; absence of brain and calvarium; incompatible with life
↑ AFP in maternal serum and amniotic fluid
Spina bifida occulta
Caudal (posterior)
Failure of vertebral arch fusion; tuft of hair or dimple over defect; usually asymptomatic
Normal AFP
Meningocele
Caudal
Meninges herniate through vertebral defect; no neural tissue involved
↑ AFP
Myelomeningocele
Caudal
Meninges and spinal cord herniate; most common clinically significant NTD; associated with Arnold-Chiari II malformation
↑↑ AFP
Figure 10 — Types of Spina Bifida. Spectrum of neural tube defects from mild (spina bifida occulta with intact skin) to severe (myelomeningocele with herniation of both meninges and spinal cord through the vertebral defect).
Folic Acid & NTD Prevention
Periconceptional folic acid supplementation (400 μg/day for low-risk; 4 mg/day for prior NTD) reduces NTD risk by 50–70%. Folic acid is essential for DNA synthesis and methylation reactions during rapid cell division. Antifolate drugs (methotrexate, trimethoprim) and anticonvulsants (valproic acid, carbamazepine) increase NTD risk.
08 Ectoderm Derivatives
The ectoderm gives rise to the nervous system, epidermis, and associated structures. It subdivides into surface ectoderm, neuroectoderm (neural tube), and neural crest.
Figure 11 — Germ Layer Derivatives in Detail. The three embryonic germ layers and their specific organ and tissue derivatives. The ectoderm produces the nervous system and epidermis, the mesoderm produces musculoskeletal and cardiovascular structures, and the endoderm produces the epithelial linings of internal organs.
NTDs; primary brain vesicles → adult brain regions
Retina (and optic nerve)
Retinoblastoma; optic cup from diencephalon outgrowth
Posterior pituitary (neurohypophysis)
Stores ADH and oxytocin from hypothalamus
Pineal gland
Melatonin secretion
Oligodendrocytes, astrocytes, ependymal cells
Microglia are mesodermal (myeloid/bone marrow origin)
Microglia are the only CNS cells NOT derived from neuroectoderm — they originate from mesodermal yolk sac macrophage precursors and colonize the developing brain. This is a commonly tested fact.
Surface ectoderm (lens placode induced by optic vesicle)
Corneal epithelium
Surface ectoderm
Corneal stroma (endothelium)
Neural crest
Sclera, choroid, ciliary body muscles
Mesoderm + neural crest
Extraocular muscles
Mesoderm (preotic somites)
Congenital cataracts may result from rubella infection during the first trimester or from galactosemia (galactitol accumulates in the lens). Coloboma (keyhole-shaped pupil) results from failure of the choroid fissure to close. Retinoblastoma arises from retinal neuroectoderm; caused by mutation in the RB1 tumor suppressor gene (chromosome 13q14).
Ear Development
Part
Embryologic Origin
External ear (pinna)
1st and 2nd pharyngeal arch mesenchyme (hillocks of His)
The mesoderm is the middle germ layer and gives rise to the musculoskeletal, cardiovascular, urogenital, and hematopoietic systems. It subdivides into paraxial, intermediate, and lateral plate mesoderm.
Paraxial mesoderm segments into ~42–44 somite pairs in a cranial-to-caudal sequence. Somitogenesis is controlled by a segmentation clock involving Notch and Wnt oscillations. Each somite differentiates into: sclerotome (ventromedial, induced by SHH from notochord) → vertebrae and ribs; dermomyotome → dermatome (dermis of back) + myotome (skeletal muscle). The number of somite pairs correlates with embryonic age.
10 Endoderm Derivatives
The endoderm lines the primitive gut tube and gives rise to the epithelial lining of the GI tract, respiratory tract, and associated glandular organs. Remember: endoderm forms the parenchyma (functional epithelium) while mesoderm forms the surrounding stroma, smooth muscle, and vasculature.
Endoderm Derivative Table
System
Endoderm-Derived Structures
GI epithelium
Epithelial lining from pharynx to rectum (above pectinate line)
Liver
Hepatocytes, biliary epithelium (from hepatic diverticulum of foregut)
Pancreas
Exocrine and endocrine pancreas (from dorsal and ventral pancreatic buds)
Thyroid
Follicular cells (from foramen cecum of tongue); C cells from neural crest
Parathyroid
Chief and oxyphil cells (3rd and 4th pharyngeal pouches)
Thymus
Epithelial component (3rd pharyngeal pouch); lymphocytes are mesodermal
Respiratory
Epithelial lining of larynx, trachea, bronchi, alveoli
The thyroid gland descends from the foramen cecum at the base of the tongue via the thyroglossal duct. Remnants can form thyroglossal duct cysts (midline neck mass that elevates with swallowing and tongue protrusion) or lingual thyroid (ectopic thyroid tissue at the base of the tongue).
Endoderm vs. Mesoderm — Key Distinctions
A common source of confusion is which portions of organs are endodermal vs. mesodermal. The general rule: endoderm forms the epithelial lining and glandular parenchyma, while mesoderm forms the surrounding connective tissue, smooth muscle, and blood vessels.
Follicular cells (endoderm); C cells (neural crest)
Capsule, vasculature, connective tissue
11 Neural Crest Derivatives
The neural crest is often called the "fourth germ layer" because of its extraordinary diversity of derivatives. Neural crest cells originate at the neural tube-ectoderm junction and migrate extensively throughout the embryo.
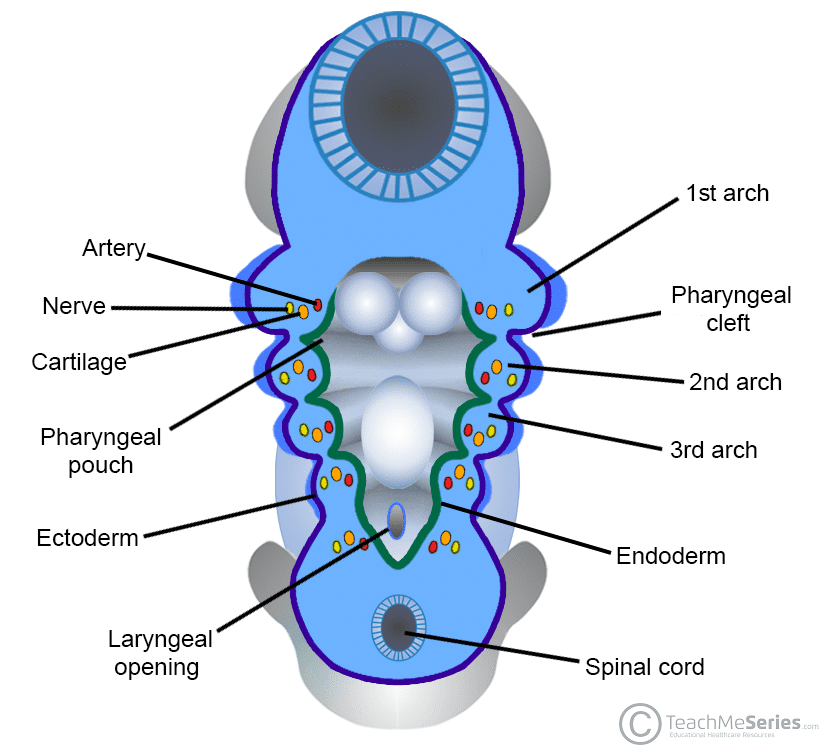
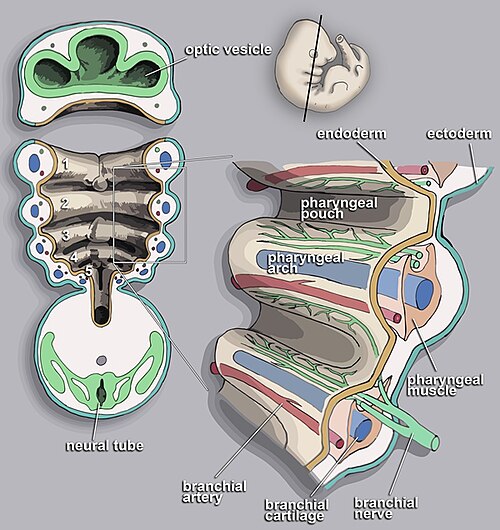
The pharyngeal (branchial) apparatus consists of arches, pouches, clefts, and membranes. Six arches develop (the 5th regresses), each containing mesoderm-derived muscle, neural crest-derived cartilage/bone, an aortic arch artery, and a cranial nerve. Understanding arch derivatives is essential for explaining head and neck anatomy and congenital anomalies.
Figure 12 — The Pharyngeal Apparatus. Overview of the pharyngeal arches, pouches (endoderm-lined, internal), and clefts (ectoderm-lined, external). Each arch contains a cranial nerve, aortic arch artery, cartilage bar, and muscular component.
Pharyngeal Arch Derivatives
Arch
CN
Muscles
Skeletal
Artery
1st
V (trigeminal)
Muscles of mastication, mylohyoid, anterior digastric, tensor tympani, tensor veli palatini
Mandible (Meckel cartilage), malleus, incus, maxilla, zygomatic bone
Maxillary artery
2nd
VII (facial)
Muscles of facial expression, stapedius, stylohyoid, posterior digastric
Stapes, styloid process, lesser horn of hyoid, upper body of hyoid (Reichert cartilage)
Stapedial artery (mostly regresses)
3rd
IX (glossopharyngeal)
Stylopharyngeus (only muscle of 3rd arch)
Greater horn and lower body of hyoid
Common and internal carotid arteries
4th
X (superior laryngeal branch)
Cricothyroid, pharyngeal constrictors, levator veli palatini
Thyroid cartilage, epiglottis
Left: aortic arch. Right: proximal right subclavian
6th
X (recurrent laryngeal branch)
All intrinsic laryngeal muscles except cricothyroid
Cricoid, arytenoid, corniculate cartilages
Pulmonary arteries; left: ductus arteriosus
Figure 13 — Pharyngeal Arch Derivatives. Each pharyngeal arch gives rise to specific muscles, skeletal elements, and is innervated by a designated cranial nerve. The 1st arch (CN V) forms masticatory structures, the 2nd (CN VII) forms facial expression muscles, the 3rd (CN IX) forms the stylopharyngeus, and arches 4 and 6 (CN X) form laryngeal structures.
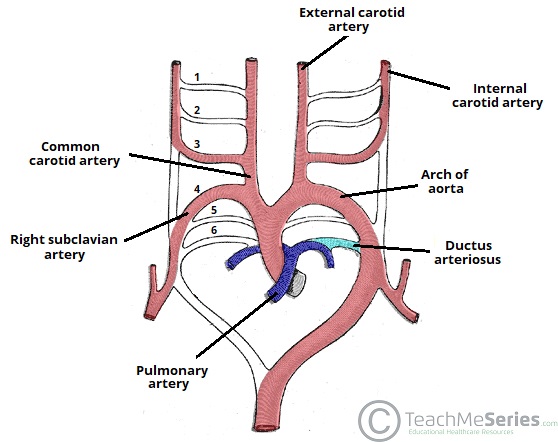
The recurrent laryngeal nerve loops under the 6th arch artery on the left (ductus arteriosus / ligamentum arteriosum) and 4th arch artery on the right (subclavian). The left recurrent laryngeal nerve has a longer course, making it more vulnerable to injury from mediastinal tumors, aortic aneurysm, or thyroid surgery.
13 Pharyngeal Pouches, Clefts & Membranes
Pharyngeal pouches are endoderm-lined outpocketings between arches on the internal (pharyngeal) side. Clefts are ectoderm-lined grooves on the external surface. Membranes are where pouch endoderm meets cleft ectoderm.
Variable position (descend with thymus); ectopic parathyroids found in anterior mediastinum
3rd pouch (ventral)
Thymus
DiGeorge: absent thymus → T-cell deficiency
4th pouch (dorsal)
Superior parathyroid glands
More consistent position than inferior parathyroids
4th pouch (ventral)
Ultimobranchial body → parafollicular C cells
Calcitonin; medullary thyroid carcinoma (MEN 2)
3rd Pouch — The Great Descent
The 3rd pouch derivatives (inferior parathyroids and thymus) descend further than 4th pouch derivatives (superior parathyroids). This means the inferior parathyroids end up below the superior parathyroids — they "overshoot" during migration. This explains why ectopic inferior parathyroids can be found in the anterior mediastinum (within or near the thymus), whereas ectopic superior parathyroids tend to be near the posterior thyroid. Surgeons must know these embryologic migration patterns to locate ectopic glands.
Pharyngeal Clefts
1st cleft → external auditory meatus (only cleft that persists)
2nd–4th clefts are obliterated by overgrowth of the 2nd arch (forming the cervical sinus)
Failure of obliteration → branchial cleft cyst (lateral neck mass anterior to sternocleidomastoid, usually 2nd cleft origin)
Figure 14 — Branchial Cleft Cyst. Clinical photograph showing a branchial cleft cyst, a lateral neck mass anterior to the sternocleidomastoid muscle. This results from failure of obliteration of the 2nd pharyngeal cleft during embryonic development.
Pharyngeal Membrane
1st membrane → tympanic membrane (the only persistent membrane)
Composed of all three layers: ectoderm (external), mesoderm (middle), endoderm (internal)
14 Heart Development & Septation
The heart is the first functional organ, with beating beginning around day 22. It develops from splanchnic lateral plate mesoderm. Cardiac development involves tube formation, looping, septation, and valve formation, with defects in any step producing congenital heart disease (CHD), the most common class of birth defects (~1% of live births).
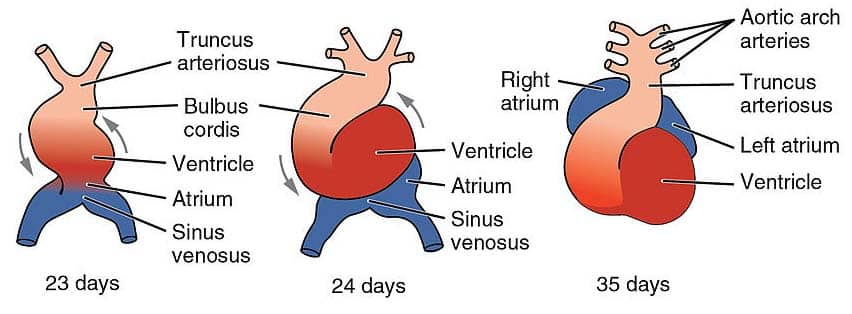
Figure 15 — Heart Tube Looping. The straight heart tube undergoes rightward looping (D-loop) beginning around day 23. This brings the primitive ventricle anteriorly and the primitive atrium posteriorly, establishing the spatial relationships necessary for normal cardiac septation.
Heart Tube Formation & Looping
Paired endocardial tubes fuse to form a single heart tube
Cardiac looping (day 23): the tube loops to the right (D-loop, dextro-loop), bringing the ventricle anterior and the atrium posterior
Abnormal looping to the left (L-loop) → dextrocardia (or situs inversus if complete)
Heart Tube Derivatives
Embryonic Structure
Adult Derivative
Truncus arteriosus
Ascending aorta, pulmonary trunk
Bulbus cordis
Smooth outflow tracts (conus arteriosus of RV, aortic vestibule of LV)
Primitive ventricle
Trabeculated portions of left and right ventricles
Primitive atrium
Trabeculated portions of left and right atria (pectinate muscles)
Sinus venosus (right horn)
Smooth part of right atrium (sinus venarum), coronary sinus, SA node
Sinus venosus (left horn)
Coronary sinus, oblique vein of left atrium
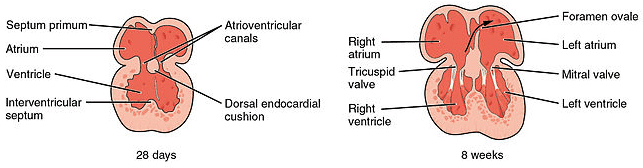
Figure 16 — Cardiac Septation. Division of the heart into four chambers involves atrial septation (septum primum and septum secundum forming the foramen ovale), ventricular septation (muscular and membranous components), and aorticopulmonary septation by neural crest-derived cells.
Septation
Septum
Mechanism
Defect
Atrial (septum primum + septum secundum)
Septum primum grows toward endocardial cushions; foramen primum closes, foramen secundum opens; septum secundum forms foramen ovale
ASD: ostium secundum (most common), ostium primum (endocardial cushion defect), patent foramen ovale
Ventricular (muscular + membranous)
Muscular septum grows upward; membranous septum formed by endocardial cushions + aorticopulmonary septum fusion
VSD: most common CHD overall; membranous VSD most common type
Aorticopulmonary (conotruncal)
Neural crest cells form spiral septum dividing truncus arteriosus into aorta and pulmonary trunk
Transposition of great vessels (failure to spiral), persistent truncus arteriosus (failure to form), tetralogy of Fallot
Tetralogy of Fallot
The most common cyanotic CHD. Results from anterosuperior displacement of the aorticopulmonary septum: (1) pulmonary infundibular stenosis, (2) overriding aorta, (3) VSD (membranous), (4) right ventricular hypertrophy (compensatory). Children present with "tet spells" (cyanotic episodes relieved by squatting, which increases SVR and reduces right-to-left shunting).
Endocardial cushion defects are associated with Down syndrome (trisomy 21). The endocardial cushions divide the atrioventricular canal into left and right AV orifices and contribute to the atrial and ventricular septa. Defects cause a common AV canal (atrioventricular septal defect).
Figure 17 — Aortic Arch Derivatives. The six pairs of aortic arch arteries undergo selective regression and remodeling to form the adult great vessels. The 3rd arches form the common and internal carotid arteries, the 4th arch forms the aortic arch (left) and proximal subclavian (right), and the 6th arch forms the pulmonary arteries and ductus arteriosus.
Aortic Arch Derivatives
Arch Artery
Left Side Derivative
Right Side Derivative
1st
Maxillary arteries (part of)
2nd
Stapedial arteries and hyoid arteries (mostly regress)
3rd
Common carotid, proximal internal carotid
Common carotid, proximal internal carotid
4th
Aortic arch (between left common carotid and left subclavian)
Proximal right subclavian artery
6th
Ductus arteriosus + left pulmonary artery
Right pulmonary artery (distal part regresses)
Valve Development
AV valves (mitral, tricuspid): develop from endocardial cushion tissue and ventricular myocardium through a process of undermining and thinning. Semilunar valves (aortic, pulmonary): develop from three swellings of subendocardial tissue in the outflow tract. Bicuspid aortic valve (~1–2% of the population) results from fusion of two of the three cusps and is the most common congenital valve anomaly; associated with coarctation of aorta and aortic aneurysm/dissection. Ebstein anomaly: failure of tricuspid valve leaflets to delaminate from ventricular wall → downward displacement into RV; associated with maternal lithium use.
Right-to-Left vs. Left-to-Right Shunts
Shunt Type
Defects
Presentation
Left-to-right (acyanotic; "late cyanosis")
VSD, ASD, PDA
Initially acyanotic; increased pulmonary blood flow → pulmonary hypertension → Eisenmenger syndrome (shunt reversal to right-to-left = late cyanosis)
Right-to-left (cyanotic; "early cyanosis")
Tetralogy of Fallot, transposition, truncus arteriosus, tricuspid atresia, total anomalous pulmonary venous return
Cyanosis from birth or early infancy; deoxygenated blood enters systemic circulation ("5 T's" of cyanotic heart disease)
15 Fetal Circulation & Shunts
Fetal circulation is adapted for gas exchange at the placenta rather than the lungs. Three critical shunts divert blood away from the non-functional fetal lungs and liver.
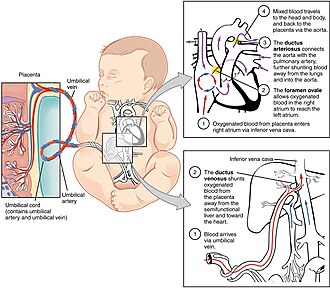
Figure 18 — Fetal Circulation. The three fetal shunts bypass the non-functional lungs and liver: the ductus venosus shunts oxygenated blood past the liver, the foramen ovale allows right-to-left atrial flow, and the ductus arteriosus diverts blood from the pulmonary trunk to the aorta.
Fetal Shunts
Shunt
Function
Postnatal Remnant
Ductus venosus
Shunts oxygenated blood from umbilical vein past the liver to IVC
Ligamentum venosum
Foramen ovale
Right-to-left atrial shunt; diverts oxygenated blood from RA to LA, bypassing lungs
Fossa ovalis (closes functionally with first breath as LA pressure exceeds RA pressure)
Ductus arteriosus
Shunts blood from pulmonary trunk to aorta, bypassing lungs
Ligamentum arteriosum
Fetal Vessel Oxygen Content
Vessel
Oxygen Content
Postnatal Remnant
Umbilical vein (single)
Highest O2 in fetus
Ligamentum teres hepatis (round ligament)
Umbilical arteries (paired)
Low O2 (return deoxygenated blood to placenta)
Medial umbilical ligaments
Allantois / urachus
N/A
Median umbilical ligament
Closure of Fetal Shunts
Ductus arteriosus closes in response to increased O2 and decreased prostaglandin E2 (PGE2) after birth. Indomethacin (prostaglandin inhibitor) promotes closure in premature infants with patent ductus arteriosus (PDA). Conversely, PGE1 (alprostadil) keeps the DA open in ductal-dependent lesions (e.g., transposition of great vessels, critical coarctation) to maintain systemic perfusion until surgical repair.
Patent ductus arteriosus (PDA) produces a continuous "machinery" murmur at the left upper sternal border. It is associated with prematurity and congenital rubella. The ductus normally closes within 24–48 hours of birth.
16 Respiratory System Development
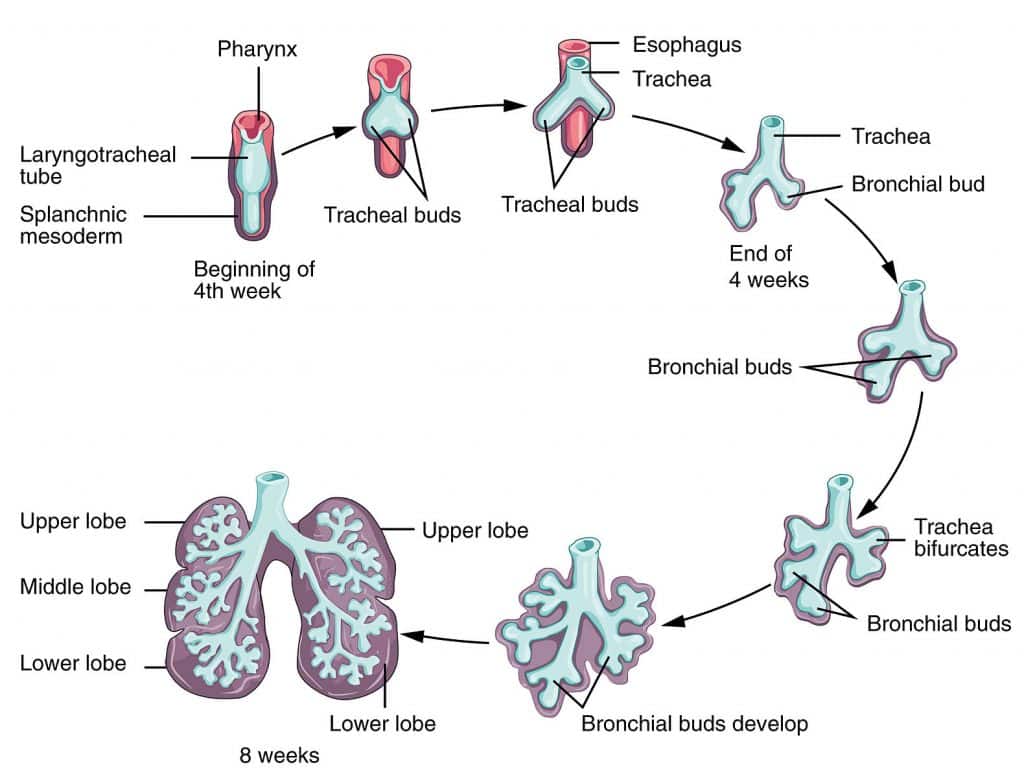
The respiratory system develops from a ventral outgrowth of the foregut endoderm (the laryngotracheal diverticulum) during week 4. The epithelium is endoderm-derived; cartilage, smooth muscle, and vasculature are mesoderm-derived.
Figure 20 — Respiratory System Development. The respiratory system develops from a ventral outgrowth (laryngotracheal diverticulum) of the foregut. The tracheoesophageal septum separates the trachea anteriorly from the esophagus posteriorly, and the bronchial buds branch to form the bronchial tree.
Branching morphogenesis (up to terminal bronchioles); resembles a gland; not viable if born
Canalicular
Weeks 16–26
Respiratory bronchioles and primitive alveoli form; capillaries approach airspaces; type II pneumocytes begin to appear; viability possible at ~24 weeks
Saccular (terminal sac)
Weeks 26–36
Terminal sacs (primitive alveoli) form; type I and type II pneumocytes mature; surfactant production increases
Alveolar
Week 36 → age 8 years
Mature alveoli with thin gas-exchange barrier; alveolar number continues to increase postnatally
Surfactant & Neonatal Respiratory Distress
Type II pneumocytes produce surfactant (dipalmitoylphosphatidylcholine / DPPC) starting ~week 24, with adequate levels by ~week 35. Surfactant reduces alveolar surface tension, preventing atelectasis. Prematurity → insufficient surfactant → neonatal respiratory distress syndrome (NRDS). The lecithin:sphingomyelin (L:S) ratio in amniotic fluid assesses lung maturity (L:S ≥ 2:1 = mature). Maternal corticosteroids (betamethasone) given 24–48 hours before preterm delivery stimulate fetal surfactant production.
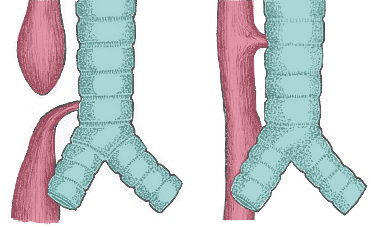
Figure 21 — Types of Tracheoesophageal Fistula. The most common type (~85%) is esophageal atresia with distal TEF, where the upper esophagus ends in a blind pouch while the lower esophagus communicates with the trachea. Other variants include H-type fistula and isolated esophageal atresia.
Tracheoesophageal fistula (TEF) results from abnormal partitioning of the foregut by the tracheoesophageal septum. The most common type (~85%) is esophageal atresia with distal TEF: the upper esophagus ends in a blind pouch while the lower esophagus communicates with the trachea. Presents with polyhydramnios, drooling, choking with first feed, and inability to pass an NG tube.
17 GI Development: Foregut, Midgut & Hindgut
The primitive gut tube forms by cranial-caudal and lateral folding of the embryonic disc, incorporating the yolk sac endoderm. The gut is divided into foregut, midgut, and hindgut based on blood supply from the three ventral branches of the aorta.
Gut Tube Divisions
Division
Blood Supply
Structures
Foregut
Celiac trunk
Pharynx to proximal duodenum (to ampulla of Vater); liver, gallbladder, pancreas, spleen (mesodermal but foregut mesentery)
Midgut
Superior mesenteric artery (SMA)
Distal duodenum to proximal 2/3 of transverse colon
Hindgut
Inferior mesenteric artery (IMA)
Distal 1/3 of transverse colon to upper anal canal (above pectinate line)
Midgut Rotation
The midgut herniates into the umbilical cord during week 6 (physiologic herniation) and rotates 270° counterclockwise around the SMA axis before returning to the abdominal cavity by week 10.
Abnormality
Mechanism
Clinical Presentation
Omphalocele
Failure of midgut to return to abdominal cavity; covered by peritoneal sac
Midline defect at umbilicus; associated with trisomies (13, 18), Beckwith-Wiedemann syndrome
Gastroschisis
Paraumbilical body wall defect (usually right of umbilicus); NOT covered by sac
Exposed bowel; not associated with chromosomal anomalies; associated with young maternal age
Malrotation
Incomplete 270° rotation; narrow mesenteric base
Risk of midgut volvulus (surgical emergency); Ladd bands may obstruct duodenum
Meckel diverticulum
Persistence of vitelline (omphalomesenteric) duct
Rule of 2s: 2% of population, 2 feet from ileocecal valve, 2 inches long; may contain ectopic gastric or pancreatic tissue → bleeding
Foregut Developmental Events
Tracheoesophageal septum divides foregut into trachea (anterior) and esophagus (posterior)
Liver bud (hepatic diverticulum) arises from ventral foregut endoderm into septum transversum mesoderm
Ventral pancreatic bud (forms uncinate process and main pancreatic duct) rotates posterior to fuse with the dorsal pancreatic bud (forms body, tail, and accessory duct)
Annular pancreas: ventral bud encircles the duodenum during rotation → duodenal obstruction
GI Atresias
Duodenal atresia: failure of recanalization of the duodenal lumen (which is initially solid); "double bubble" sign on X-ray; associated with Down syndrome. Jejunal/ileal atresia: vascular accident (ischemia) during development; "apple peel" deformity; NOT associated with chromosomal abnormalities. Colonic atresia: rare; also vascular accident.
The pectinate (dentate) line marks the junction of hindgut endoderm and ectoderm from the proctodeum. Above the line: columnar epithelium, internal hemorrhoids (painless, visceral innervation), portal venous drainage. Below the line: squamous epithelium, external hemorrhoids (painful, somatic innervation via inferior rectal nerve), systemic venous drainage (IVC).
Hindgut & Cloaca
The cloaca is the common chamber at the caudal end of the hindgut that receives the allantois anteriorly and the hindgut posteriorly. The urorectal septum divides the cloaca into the urogenital sinus (anterior) and the anorectal canal (posterior). Failure of urorectal septum formation leads to persistent cloaca or rectourethral/rectovaginal fistulas.
Liver & Biliary Development
Structure
Origin
Clinical Correlate
Hepatic diverticulum (liver bud)
Ventral foregut endoderm
Hepatocytes, intrahepatic bile ducts
Gallbladder and cystic duct
Caudal portion of hepatic diverticulum
Biliary atresia (obliteration of extrahepatic bile ducts; most common indication for pediatric liver transplant)
Hepatic resident macrophages; NOT endoderm-derived
Spleen Development
The spleen develops within the dorsal mesogastrium from mesoderm (NOT endoderm, despite its foregut location). It is the only solid intraperitoneal organ that is entirely mesodermal. Accessory spleens (~10% of population) are found near the splenic hilum or in the greater omentum and are clinically relevant after splenectomy for hematologic conditions (e.g., ITP) since they can hypertrophy and maintain splenic function.
18 Renal System Development
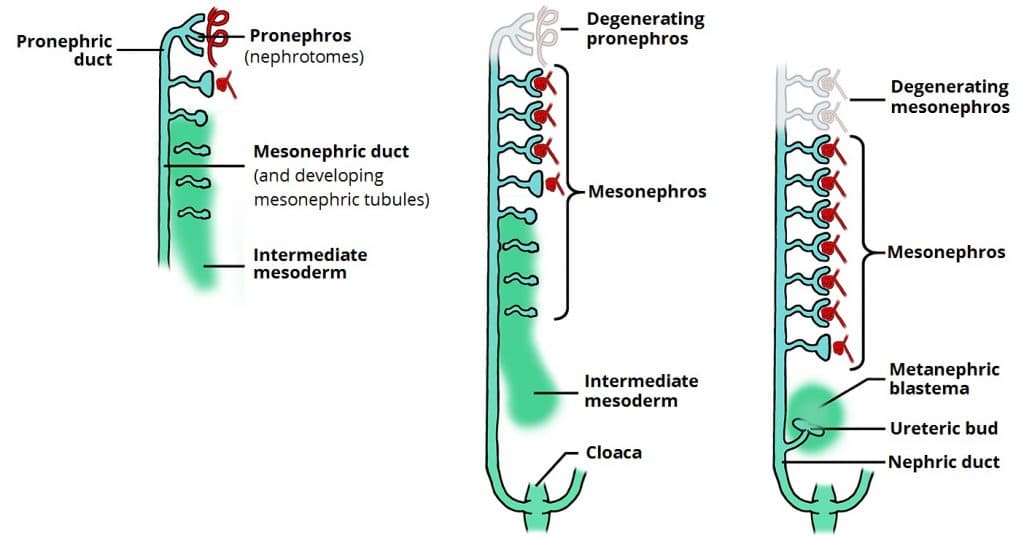
The urinary system develops from intermediate mesoderm in a cranial-to-caudal sequence through three successive kidney systems. The definitive kidney (metanephros) is derived from two sources: the metanephric mesoderm (mesenchyme) and the ureteric bud.
Figure 22 — Kidney Development Stages. Three successive kidney systems develop in a cranial-to-caudal sequence: the pronephros (nonfunctional, degenerates), mesonephros (interim kidney, duct persists as Wolffian duct), and metanephros (definitive kidney from ureteric bud and metanephric mesenchyme).
Three Kidney Systems
System
Timing
Fate
Pronephros
Week 4
Nonfunctional; degenerates; pronephric duct persists as mesonephric duct
Mesonephros
Weeks 4–8
Functions briefly as interim kidney; mesonephric (Wolffian) duct becomes vas deferens, epididymis, seminal vesicles in males
Metanephros
Week 5 onward
Definitive kidney; ureteric bud (from mesonephric duct) forms collecting system; metanephric mesenchyme forms nephrons
Metanephric mesenchyme (blastema) → glomerulus, Bowman capsule, proximal and distal tubules, loop of Henle
Reciprocal induction: ureteric bud signals mesenchyme to form nephrons; mesenchyme signals ureteric bud to branch
Kidneys ascend from the pelvis to the lumbar region, acquiring new arterial supply at each level
Congenital Renal Anomalies
Anomaly
Embryologic Basis
Clinical Significance
Horseshoe kidney
Lower poles fuse during ascent; trapped under IMA
Usually asymptomatic; increased risk of Wilms tumor; associated with Turner syndrome
Pelvic kidney
Failure to ascend
Usually asymptomatic; vulnerable to trauma
Unilateral renal agenesis
Failure of ureteric bud to develop or contact mesenchyme
Compatible with life; contralateral kidney hypertrophies
Bilateral renal agenesis (Potter sequence)
Both ureteric buds fail
Oligohydramnios → pulmonary hypoplasia, limb deformities, flattened facies; incompatible with life
Duplex collecting system
Early splitting of ureteric bud
Double ureters; increased UTI risk
Multicystic dysplastic kidney
Abnormal ureteric bud-mesenchyme interaction
Nonfunctional kidney; most common renal cystic disease in children
Potter sequence (oligohydramnios sequence) results from any cause of severely decreased amniotic fluid (bilateral renal agenesis, posterior urethral valves, bilateral multicystic kidneys). The classic features are pulmonary hypoplasia (usually cause of death), flattened facies, limb contractures, and growth restriction.
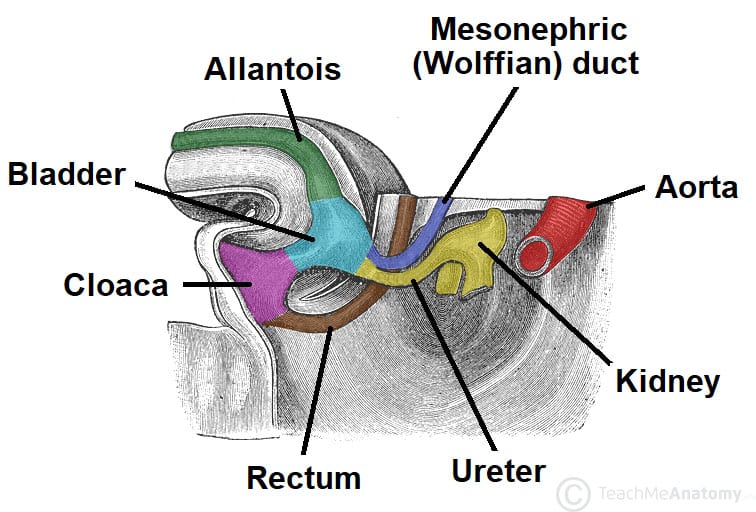
Figure 23 — Bladder and Ureteric Development. The bladder develops from the upper urogenital sinus, with the ureters (from the ureteric bud) and mesonephric ducts (Wolffian ducts) establishing their definitive connections. The trigone of the bladder is formed by incorporation of mesonephric duct tissue.
Bladder & Urogenital Sinus Development
The urogenital sinus is the anterior division of the cloaca (after the urorectal septum divides it from the anorectal canal). It differentiates into three regions:
The allantois connects the bladder to the umbilicus. Its lumen normally obliterates to form the urachus, which becomes the median umbilical ligament. A patent urachus results in drainage of urine from the umbilicus. A urachal cyst occurs when only a segment remains patent, presenting as an infected midline infraumbilical mass.
19 Genital System & Sexual Differentiation
The genital system develops from the same precursor structures in both sexes. Sex determination depends on the presence or absence of the SRY gene on the Y chromosome. Until week 6, male and female embryos are phenotypically indistinguishable (indifferent gonad stage).
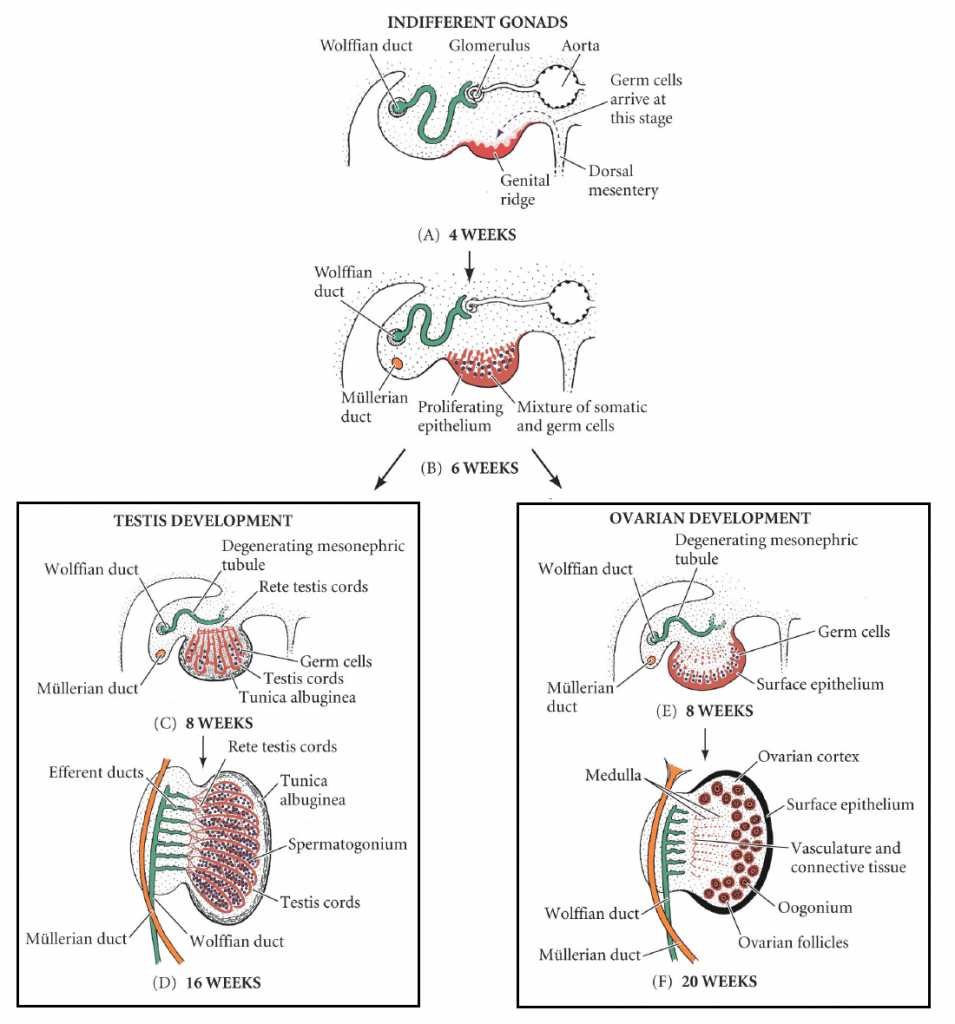
Figure 24 — Gonadal Differentiation. The indifferent gonad differentiates into a testis (driven by SRY gene on the Y chromosome) or ovary (default pathway in XX individuals). Sertoli cells produce AMH to regress Müllerian ducts, while Leydig cells produce testosterone to maintain Wolffian ducts.
Sexual Differentiation Pathway
Factor
Source
Effect
SRY gene
Y chromosome
Induces Sertoli cell differentiation in gonadal ridge → testis development
Testosterone
Leydig cells (testis)
Stimulates Wolffian (mesonephric) duct → epididymis, vas deferens, seminal vesicles
5α-reductase deficiency: XY, normal internal male structures (testosterone-dependent), ambiguous or female external genitalia at birth (DHT-dependent); virilization at puberty. Androgen insensitivity syndrome (AIS): XY, defective androgen receptor; testes present (may be cryptorchid), female external genitalia, absent uterus (AMH still functions); presents as primary amenorrhea. Congenital adrenal hyperplasia (21-hydroxylase deficiency): XX, excess adrenal androgens; virilized female genitalia at birth with salt-wasting crisis.
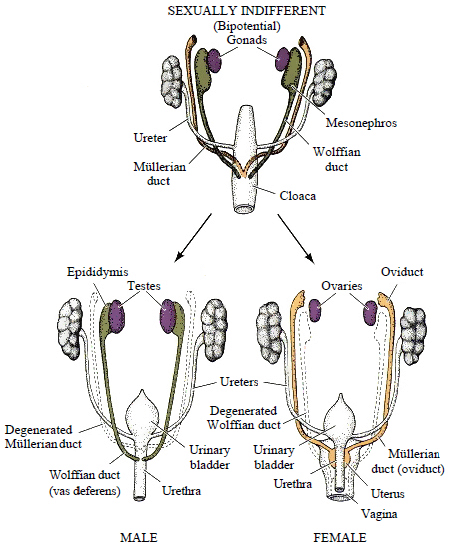
Figure 25 — Internal Genital Duct Development. In males, testosterone maintains the Wolffian (mesonephric) ducts which form the epididymis, vas deferens, and seminal vesicles, while AMH causes Müllerian duct regression. In females, the Müllerian (paramesonephric) ducts persist to form the uterine tubes, uterus, and upper vagina.
Duct System Development
Duct
Present In
Fate in Male
Fate in Female
Mesonephric (Wolffian) duct
Both sexes initially
Epididymis, vas deferens, seminal vesicles, ejaculatory duct
Degenerates (remnants: Gartner duct cyst in vaginal wall)
Paramesonephric (Müllerian) duct
Both sexes initially
Degenerates due to AMH (remnant: appendix testis)
Uterine tubes, uterus, upper 1/3 of vagina
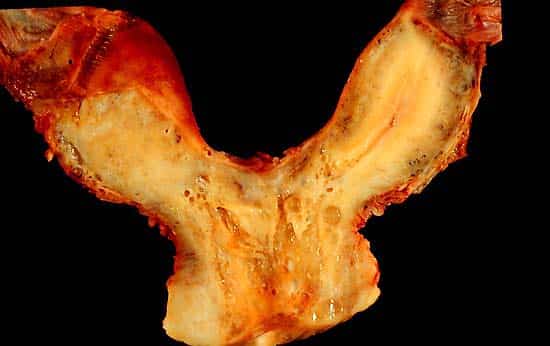
Figure 26 — Müllerian Duct Anomaly: Bicornuate Uterus. Incomplete fusion of the Müllerian ducts results in a bicornuate (heart-shaped) uterus with two uterine horns. This is one of several anomalies along a spectrum from septate uterus (mildest) to uterus didelphys (complete non-fusion).
Uterine Anomalies from Müllerian Duct Defects
Anomaly
Mechanism
Clinical Significance
Uterus didelphys
Complete failure of Müllerian duct fusion
Two separate uteri and cervices; may have double vagina
Bicornuate uterus
Incomplete fusion of Müllerian ducts
Heart-shaped uterus; increased risk of preterm labor, malpresentation
Septate uterus
Failure of resorption of the uterovaginal septum after fusion
Most common uterine anomaly; highest risk of recurrent miscarriage; can be surgically corrected
Unicornuate uterus
Agenesis of one Müllerian duct
Single uterine horn; increased ectopic pregnancy risk
Müllerian agenesis (MRKH syndrome)
Failure of Müllerian duct development; 46,XX
Absent uterus and upper vagina; normal ovaries and secondary sexual characteristics; primary amenorrhea
Gonadal Descent
The gubernaculum guides testicular descent from the posterior abdominal wall through the inguinal canal into the scrotum, pulling a sleeve of peritoneum (processus vaginalis). The processus vaginalis normally obliterates; failure causes indirect inguinal hernia or communicating hydrocele. Cryptorchidism (undescended testis) affects ~3% of full-term males and increases the risk of infertility and testicular germ cell tumors.
In females, the gubernaculum becomes the ovarian ligament (connects ovary to uterus) and the round ligament of the uterus (passes through the inguinal canal to the labia majora). The round ligament is the homologue of the male gubernaculum. During pregnancy, the round ligament stretches and can cause round ligament pain in the groin.
20 CNS Development
The CNS develops from the neural tube, which forms three primary brain vesicles during week 4 that further subdivide into five secondary vesicles by week 5.
Small posterior fossa; cerebellar tonsils herniate through foramen magnum
Associated with myelomeningocele; hydrocephalus; syringomyelia
Dandy-Walker malformation
Failure of cerebellar vermis development; cystic dilation of 4th ventricle
Enlarged posterior fossa, hydrocephalus
Hydrocephalus
Obstruction of CSF flow (most commonly at cerebral aqueduct — aqueductal stenosis)
Enlarged head, bulging fontanelles, sunset eyes in infants
Syringomyelia
Fluid-filled cavity in central spinal cord (often cervical)
Cape-like loss of pain/temperature (spinothalamic fibers crossing at affected level); associated with Chiari I malformation
Holoprosencephaly is associated with trisomy 13 (Patau syndrome) and Sonic Hedgehog (SHH) mutations. The saying "the face predicts the brain" refers to the spectrum of midline facial defects (cyclopia, proboscis, cleft lip/palate) that correlate with the severity of forebrain malformation.
Spinal Cord Development
Zone
Inducing Signal
Derivatives
Floor plate
SHH from notochord
Ventral midline glial cells; axon guidance center
Basal plate (ventral)
SHH gradient
Motor neurons (ventral horn); motor = ventral
Alar plate (dorsal)
BMP/Wnt from roof plate
Sensory neurons (dorsal horn); sensory = dorsal
Roof plate
BMP, Wnt
Dorsal midline; commissural axon guidance
The distinction between the basal (motor) and alar (sensory) plates is maintained throughout the CNS. In the brainstem, motor nuclei are medial and sensory nuclei are lateral (the sulcus limitans separates them on the floor of the 4th ventricle). This principle explains the organization of cranial nerve nuclei.
Ventricular System & Choroid Plexus
The ventricular system develops from the lumen of the neural tube. The choroid plexus (ependymal cells + vascularized pia mater) produces CSF in all four ventricles. CSF flows: lateral ventricles → interventricular foramina (of Monro) → 3rd ventricle → cerebral aqueduct (of Sylvius) → 4th ventricle → foramina of Luschka (lateral, 2) and Magendie (median, 1) → subarachnoid space → arachnoid granulations → dural venous sinuses. Obstruction at the cerebral aqueduct (aqueductal stenosis) is the most common cause of congenital hydrocephalus.
21 Musculoskeletal & Limb Development
Limb buds appear during weeks 4–5, with upper limbs developing slightly before lower limbs. Three signaling centers coordinate limb patterning along three axes.
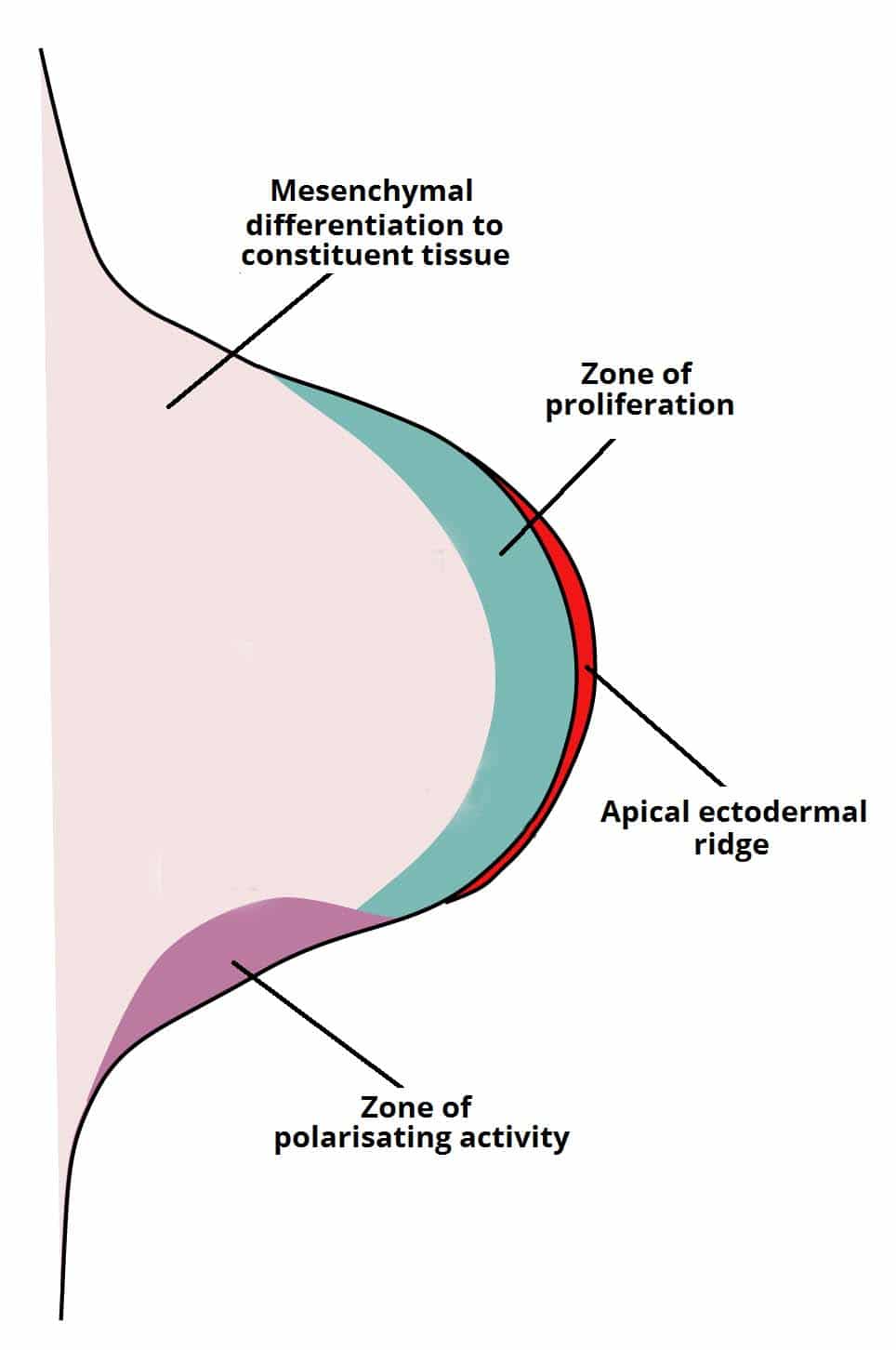
Figure 28 — Limb Bud and Apical Ectodermal Ridge (AER). The AER at the distal tip of the limb bud secretes FGFs to drive proximal-to-distal outgrowth. The zone of polarizing activity (ZPA) at the posterior margin produces SHH for anterior-posterior patterning, while Wnt7a from the dorsal ectoderm patterns the dorsal-ventral axis.
Limb Signaling Centers
Center
Signal
Axis
Defect if Lost
Apical ectodermal ridge (AER)
FGFs
Proximal-distal (shoulder → fingers)
Limb truncation (amelia, meromelia)
Zone of polarizing activity (ZPA)
SHH
Anterior-posterior (thumb → pinky)
Polydactyly, mirror-image duplication
Dorsal ectoderm
Wnt7a
Dorsal-ventral (back of hand → palm)
Dorsal-ventral axis reversal
Bone Development
Bones form by two mechanisms: endochondral ossification (cartilage model replaced by bone — long bones, vertebrae, pelvis) and intramembranous ossification (bone forms directly from mesenchyme without cartilage template — flat bones of skull, clavicle). Achondroplasia results from a gain-of-function mutation in FGFR3, which constitutively inhibits chondrocyte proliferation in the epiphyseal growth plate, causing short limbs with normal trunk.
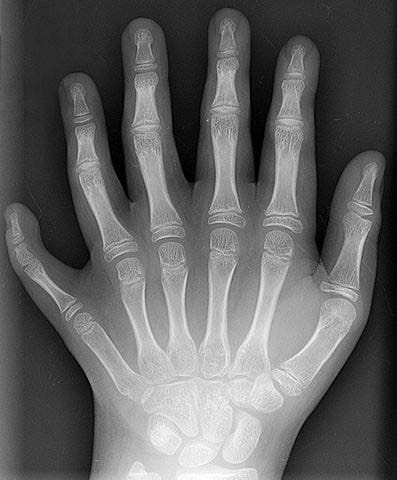
Figure 29 — Polydactyly. Radiograph demonstrating polydactyly (supernumerary digits), which can result from excess SHH signaling from the zone of polarizing activity. Polydactyly is associated with trisomy 13 (Patau syndrome) and Ellis-van Creveld syndrome.
Limb Anomalies
Anomaly
Mechanism
Association
Polydactyly
Excess SHH signaling or genetic (often autosomal dominant)
Trisomy 13, Ellis-van Creveld syndrome
Syndactyly
Failure of apoptosis between digits
Apert syndrome (FGFR2 mutation)
Amelia
Complete absence of limb; AER failure
Thalidomide exposure (phocomelia: absent proximal limb with hands/feet attached to trunk)
Clubfoot (talipes equinovarus)
Multifactorial; abnormal muscle/tendon development
Common (~1/1000 births); associated with oligohydramnios
Diaphragm Development
The diaphragm develops from four embryonic structures: (1) septum transversum (central tendon; C3–C5 innervation explains phrenic nerve origin), (2) pleuroperitoneal folds (close the pericardioperitoneal canals), (3) body wall mesoderm (peripheral muscular rim), (4) esophageal mesentery (crura).
Congenital diaphragmatic hernia (Bochdalek hernia) results from failure of the pleuroperitoneal fold to close, usually on the left posterolateral side (~90%). Abdominal contents herniate into the thorax, causing pulmonary hypoplasia. Presents at birth with respiratory distress, scaphoid abdomen, and absent breath sounds on the affected side.
Body Cavity Development
The intraembryonic coelom forms within the lateral plate mesoderm and is initially a continuous horseshoe-shaped cavity. Partitioning creates the three body cavities:
Partition
Separates
Defect
Pleuropericardial folds
Pleural cavity from pericardial cavity
Pericardial effusion into pleural space (rare)
Pleuroperitoneal folds
Pleural cavity from peritoneal cavity
Congenital diaphragmatic hernia (Bochdalek)
Septum transversum
Thoracic from abdominal cavity (becomes central tendon of diaphragm)
Absent or hypoplastic clavicles, wide fontanelles, supernumerary teeth; patients can approximate shoulders anteriorly
22 Congenital Anomalies by System
Congenital anomalies affect ~3% of live births and are a leading cause of infant mortality. Understanding the embryologic basis of each defect guides diagnosis, counseling, and surgical management.
Cardiovascular Anomalies
Defect
Embryologic Error
Key Feature
VSD
Incomplete fusion of muscular/membranous ventricular septum
Most common CHD; holosystolic murmur at left lower sternal border
ASD (secundum)
Excessive resorption of septum primum or deficient septum secundum
Fixed split S2; right heart volume overload
PDA
Failure of ductus arteriosus to close
Continuous machinery murmur; associated with prematurity, rubella
Coarctation of aorta
Abnormal involution of left 4th aortic arch or abnormal ductus tissue
Infantile (preductal, associated with Turner syndrome) vs. adult (postductal, rib notching)
Transposition of great vessels
Failure of aorticopulmonary septum to spiral
Aorta arises from RV, PA from LV; incompatible with life without mixing (PDA, VSD, or ASD)
GI Anomalies
Defect
Embryologic Error
Key Feature
Esophageal atresia / TEF
Abnormal tracheoesophageal septum formation
Polyhydramnios, inability to pass NG tube; associated with VACTERL
Pyloric stenosis
Hypertrophy of pyloric smooth muscle (postnatal)
Projectile nonbilious vomiting at 2–6 weeks; palpable "olive" mass
Hirschsprung disease
Failure of neural crest migration to distal colon
Absent ganglia in affected segment; functional obstruction; failure to pass meconium
Imperforate anus
Abnormal urorectal septum division of cloaca
Part of VACTERL association; may have fistula to urogenital tract
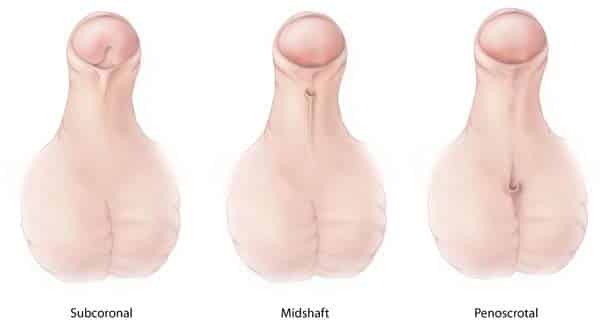
Figure 30 — Hypospadias Variants. Hypospadias results from failure of the urethral folds to fuse on the ventral surface of the penis. The urethral opening may be glanular, penile, penoscrotal, or perineal. Foreskin tissue is preserved for surgical repair (do not circumcise).
Urogenital Anomalies
Defect
Embryologic Error
Key Feature
Hypospadias
Failure of urethral folds to fuse ventrally
Urethral opening on ventral (underside) surface of penis; do NOT circumcise (foreskin used for repair)
Epispadias
Abnormal genital tubercle positioning
Urethral opening on dorsal surface; associated with bladder exstrophy
Bladder exstrophy
Failure of anterior body wall closure over the bladder
Exposed bladder mucosa on abdominal wall; associated with epispadias
Posterior urethral valves
Abnormal remnants of Wolffian duct
Most common cause of bladder outlet obstruction in male newborns; causes bilateral hydronephrosis, oligohydramnios
VACTERL Association
A non-random association of congenital defects: Vertebral anomalies, Anal atresia, Cardiac defects (VSD most common), TracheoEsophageal fistula, Renal anomalies, Limb defects (radial anomalies). Diagnosis requires at least 3 components. Not a syndrome (no single genetic cause); the etiology remains unknown.
23 Teratology & Critical Periods
A teratogen is any agent that can cause a structural or functional defect in the developing embryo or fetus. Teratogenicity depends on the timing of exposure, dose, genotype of the embryo, and the specific agent. The most vulnerable period is weeks 3–8 (organogenesis).
NSAIDs in 3rd trimester (premature closure of ductus arteriosus), high-dose aspirin
Acne
Topical erythromycin, azelaic acid
Isotretinoin (category X; iPLEDGE program required)
Principles of Teratology (Wilson Principles)
Susceptibility depends on the genotype of the embryo and its interaction with the environment
Susceptibility varies with the developmental stage at exposure
Teratogenic agents act by specific mechanisms on developing cells and tissues
The final manifestation depends on whether the access of the agent to the developing tissue occurs by dose and duration
Teratogenic effects range from death to malformation to growth retardation to functional deficits
As the dosage increases, manifestations increase in frequency and severity
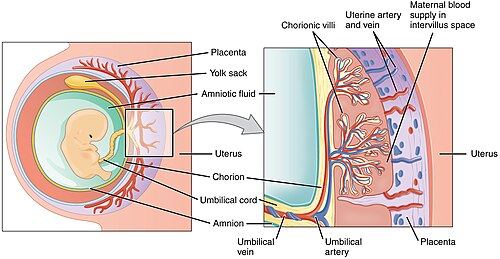
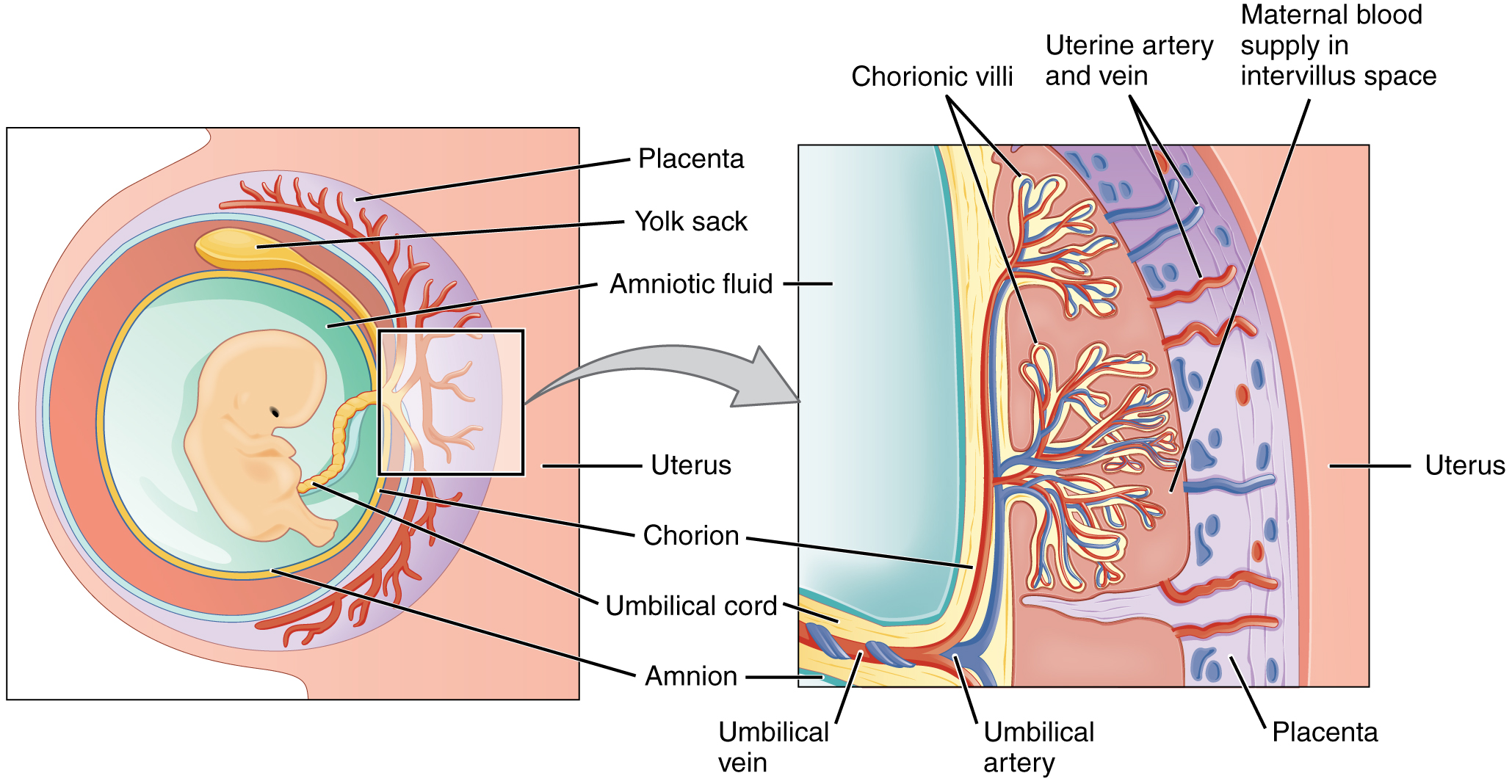
24 Placenta, Membranes & Umbilical Cord
The placenta is a fetomaternal organ that serves as the site of nutrient, gas, and waste exchange. It also functions as an endocrine organ, producing hormones essential for pregnancy maintenance.
Figure 31 — Placental Anatomy. The placenta consists of a fetal portion (chorionic villi with fetal blood vessels surrounded by syncytiotrophoblast) and a maternal portion (decidua basalis). Maternal blood fills the intervillous space, allowing exchange of gases, nutrients, and waste across the placental barrier.
Placental Structure
Component
Origin
Description
Chorionic villi (fetal side)
Trophoblast + extraembryonic mesoderm
Fetal blood vessels surrounded by syncytiotrophoblast; float in maternal blood in intervillous space
Decidua basalis (maternal side)
Endometrium
Portion of endometrium underlying the implanted embryo; forms maternal portion of placenta
Syncytiotrophoblast
Trophoblast
Multinucleated outer layer; in direct contact with maternal blood; secretes hCG, hPL, estrogen, progesterone
Cytotrophoblast
Trophoblast
Inner layer of individual cells; stem cells for syncytiotrophoblast; predominant in first trimester
Figure 32 — Placental Cross-Section. Detailed view of the placental barrier: fetal blood circulates within chorionic villi (containing fetal capillaries), which are bathed in maternal blood in the intervillous space. The syncytiotrophoblast layer mediates exchange and hormone secretion.
Placental Hormones
Hormone
Function
hCG
Maintains corpus luteum (progesterone production) in first trimester; basis for pregnancy tests
Human placental lactogen (hPL)
Promotes lipolysis and insulin resistance in mother to divert glucose to fetus; stimulates breast development
Progesterone
Maintains endometrium; suppresses uterine contractions; placenta takes over from corpus luteum by week 8–12
The umbilical cord contains two umbilical arteries (carry deoxygenated blood from fetus to placenta) and one umbilical vein (carries oxygenated blood from placenta to fetus), embedded in Wharton jelly (mucoid connective tissue). A single umbilical artery (present in ~1% of births) is associated with congenital anomalies, particularly renal and cardiac defects.
The amniotic fluid volume reflects a balance between fetal swallowing (removes fluid) and fetal urination (adds fluid). Any condition that impairs swallowing increases fluid (polyhydramnios), while any condition that impairs urine output decreases fluid (oligohydramnios).
Placental Pathology
Condition
Description
Clinical Significance
Placenta previa
Placenta implants over or near the internal cervical os
Painless vaginal bleeding in 3rd trimester; cesarean delivery indicated
~95% tubal (ampulla most common); rupture causes hemoperitoneum; treat with methotrexate or surgery
Decidual Layers
Layer
Location
Fate
Decidua basalis
Between embryo and myometrium (deep to implantation site)
Maternal component of placenta; shed at delivery
Decidua capsularis
Overlying the embryo (superficial to implantation site)
Thins and degenerates as embryo grows; fuses with decidua parietalis
Decidua parietalis
Lines the rest of the uterine cavity
Fuses with decidua capsularis; shed at delivery
25 Twinning & Multiple Gestations
Twins occur in approximately 1 in 80 pregnancies (higher with assisted reproduction). The type of twinning determines the arrangement of placental membranes and the associated risks.
Dizygotic vs. Monozygotic Twins
Feature
Dizygotic (Fraternal)
Monozygotic (Identical)
Mechanism
Two oocytes fertilized by two sperm
Single fertilized ovum splits
Frequency
~2/3 of all twins
~1/3 of all twins
Genetics
No more alike than siblings
Genetically identical
Chorionicity
Always dichorionic-diamniotic
Depends on timing of division
Figure 33 — Monozygotic Twin Membrane Arrangements. The timing of embryo splitting determines the membrane arrangement: division before day 3 produces dichorionic-diamniotic twins, days 4–8 produces monochorionic-diamniotic twins, days 8–12 produces monochorionic-monoamniotic twins, and after day 13 results in conjoined twins.
Monozygotic Twin Membrane Arrangement
Timing of Division
Membrane Type
Frequency
Days 0–3 (before morula)
Dichorionic-diamniotic (separate placentas)
~25–30%
Days 4–8 (inner cell mass splits)
Monochorionic-diamniotic (shared placenta, separate amnions)
~65–70%
Days 8–12 (after amniotic cavity forms)
Monochorionic-monoamniotic (shared placenta and amnion)
~1–2%
Days 13+ (incomplete split)
Conjoined twins
Very rare
Twin-to-Twin Transfusion Syndrome (TTTS)
Occurs in monochorionic twins with shared placental vascular anastomoses. One twin (donor) shunts blood to the other (recipient). Donor: anemia, oligohydramnios, growth restriction. Recipient: polycythemia, polyhydramnios, heart failure. Treatment: fetoscopic laser ablation of communicating placental vessels. TTTS only occurs in monochorionic placentas because dichorionic twins have separate vascular beds.
Ultrasound every 2 weeks starting at 16 weeks; MCA Doppler surveillance
Monochorionic-monoamniotic
Cord entanglement (leading cause of mortality), TTTS, prematurity
Intensive surveillance; planned delivery at 32–34 weeks
Conjoined twins
Organ sharing, surgical separation challenges
MRI for organ mapping; multidisciplinary surgical planning
The determination of chorionicity is best assessed by first-trimester ultrasound. The "twin peak" (lambda) sign indicates dichorionic placentation (triangular projection of placental tissue between the membranes). The "T-sign" (thin membrane meeting the placenta at a right angle) indicates monochorionic placentation. Chorionicity, not zygosity, determines pregnancy risk.
26 Fetal Development & Milestones
The fetal period (weeks 9–38) is characterized by rapid growth and functional maturation of organ systems established during the embryonic period.
Key Fetal Milestones
Week
Milestone
Week 9
Fetal period begins; liver is major site of hematopoiesis; external genitalia begin to differentiate
Week 10
Intestines return to abdominal cavity from physiologic herniation; kidneys begin producing urine
Week 12
External genitalia distinguishable as male or female; ossification centers in long bones; fetal movements begin
Week 14
Gender identifiable by ultrasound; lanugo hair appears
Week 16
Bone marrow begins hematopoiesis; eyes face anteriorly
Neonatal kidneys have low GFR and limited concentrating ability
Liver (glucuronidation)
Immature at birth; matures over first weeks
Physiologic jaundice of the newborn (inadequate conjugation of bilirubin)
Immune system
IgG crosses placenta (passive immunity); IgM does not
Elevated IgM in newborn suggests intrauterine infection (TORCH)
Hematopoiesis Sites Over Development
Period
Primary Site
Weeks 3–8
Yolk sac (mesoblastic period)
Weeks 6–30
Liver (and spleen) — hepatic period; liver is the major site by week 9
Week 18 onward
Bone marrow (medullary period); becomes the primary site by week 28
The sequence of hematopoiesis sites is commonly remembered as "Young Liver Synthesizes Blood" — Yolk sac → Liver → Spleen → Bone marrow. Extramedullary hematopoiesis (liver, spleen) in postnatal life indicates severe marrow stress (e.g., myelofibrosis, severe hemolytic anemias).
Fetal Hemoglobin & Oxygen Transport
Fetal hemoglobin (HbF) consists of two alpha and two gamma globin chains (α2γ2), in contrast to adult hemoglobin HbA (α2β2). HbF has a higher oxygen affinity than HbA because it binds 2,3-BPG less avidly. This left-shifted oxygen-hemoglobin dissociation curve facilitates oxygen transfer from maternal blood to fetal blood across the placenta. HbF production begins to decline before birth, and the switch to HbA is largely complete by ~6 months of age.
Hemoglobin Switching & Clinical Significance
The hemoglobin switch from gamma to beta globin chains explains why sickle cell disease and β-thalassemia do not manifest until after ~6 months of age (when HbF declines and HbA/HbS predominates). Alpha-thalassemia, however, can present in utero (Hb Barts hydrops fetalis — all four alpha genes deleted → γ4 tetramers with extremely high O2 affinity → fetal hydrops and death). Hydroxyurea induces HbF production in sickle cell patients, improving symptoms.
Fetal Weight & Growth Milestones
Gestational Age
Crown-Rump Length
Weight (approx.)
Key Features
12 weeks
~6 cm
~14 g
Gender identifiable; ossification begins
16 weeks
~12 cm
~100 g
Lanugo hair; rapid growth
20 weeks
~16 cm
~300 g
Quickening; vernix caseosa
24 weeks
~21 cm
~600 g
Surfactant production begins; limit of viability
28 weeks
~25 cm
~1000 g
Eyes open; reasonable viability
32 weeks
~28 cm
~1700 g
Subcutaneous fat; fingernails to fingertips
36 weeks
~34 cm
~2500 g
Lung maturity; firm grasp
40 weeks (term)
~36 cm
~3400 g
Full maturity
27 Clinical Correlates
Embryologic knowledge directly translates to clinical practice across multiple specialties. The following clinical scenarios demonstrate how developmental principles guide diagnosis and management.
Prenatal Screening & Diagnosis
Test
Timing
What It Detects
First trimester screen (PAPP-A + free β-hCG + nuchal translucency)
Thyroglossal duct cyst: midline neck mass that moves with swallowing and tongue protrusion; excised with Sistrunk procedure (includes the central body of the hyoid bone). Branchial cleft cyst: lateral neck mass anterior to SCM (usually 2nd cleft origin). Meckel diverticulum: true diverticulum (all three wall layers) on the antimesenteric border of the ileum; may cause painless GI bleeding in children from ectopic gastric mucosa. Undescended testis: orchiopexy recommended by age 6–12 months to reduce infertility and malignancy risk (does not eliminate cancer risk).
Genetic Counseling Applications
Advanced maternal age (≥35 years): increased risk of nondisjunction → aneuploidies (trisomy 21, 18, 13); offer screening and diagnostic testing
Advanced paternal age (≥40 years): increased risk of de novo point mutations (achondroplasia, Marfan syndrome, neurofibromatosis)
Recurrence risk: NTDs ~3–5% after one affected child; congenital heart defects ~2–5% after one affected child
Teratogen counseling: category X drugs (isotretinoin, thalidomide, warfarin, methotrexate) are absolutely contraindicated in pregnancy; women of childbearing age require pregnancy prevention programs
Congenital Infections — TORCH Mnemonic
Infection
Transmission
Classic Features
Diagnosis
Toxoplasma
Cat feces, undercooked meat
Intracranial calcifications (diffuse), chorioretinitis, hydrocephalus, ring-enhancing lesions (in immunocompromised)
Exam Focus: The highest-yield embryology topics on board examinations include: (1) germ layer derivatives — especially neural crest; (2) heart septation defects and their associations with syndromes; (3) fetal circulation shunts and their postnatal remnants; (4) pharyngeal arch/pouch derivatives; (5) teratogens and their specific effects; (6) neural tube defects and folic acid; (7) GI development anomalies (Meckel diverticulum, TEF, midgut rotation); (8) sexual differentiation disorders; (9) kidney development and Potter sequence; (10) prenatal screening interpretation (AFP, quad screen).