Genetics & Molecular Biology
DNA structure, replication, transcription, translation, Mendelian inheritance, cytogenetics, molecular diagnostics, genetic testing, cancer genetics, and every genetic principle, mutation type, and clinical syndrome across the full scope of medical genetics.
01 Overview & Scope of Medical Genetics
Medical genetics is the study of heredity, genes, and genetic variation as they relate to human health and disease. It integrates molecular biology, cytogenetics, biochemical genetics, and clinical medicine to explain how genetic factors cause, predispose to, or modify the course of disease. A working knowledge of genetics is indispensable for understanding congenital anomalies, inborn errors of metabolism, cancer, pharmacologic variability, and the growing field of precision medicine.
Approximately 3–5% of live births have a recognizable genetic disorder, and genetic factors contribute to the majority of common diseases including coronary artery disease, diabetes, and cancer. With the advent of next-generation sequencing, gene therapy, and CRISPR-based therapeutics, genetics has moved from a diagnostic discipline to a therapeutic one. Every physician — regardless of specialty — must understand inheritance patterns, genetic testing, and the ethical implications of genomic medicine.
Core Domains of Medical Genetics
| Domain | Focus | Clinical Examples |
|---|---|---|
| Molecular Genetics | DNA structure, gene expression, mutation | Sickle cell disease, cystic fibrosis |
| Cytogenetics | Chromosome structure and number | Down syndrome, Turner syndrome |
| Biochemical Genetics | Enzyme deficiencies and metabolic pathways | Phenylketonuria, Gaucher disease |
| Clinical Genetics | Diagnosis, counseling, management of genetic disorders | Genetic counseling for BRCA carriers |
| Cancer Genetics | Oncogenes, tumor suppressors, hereditary syndromes | Li–Fraumeni syndrome, Lynch syndrome |
| Pharmacogenomics | Genetic variation in drug response | CYP2D6 polymorphisms, HLA-B*5701 testing |
02 Key Terminology & Abbreviations
| Term / Abbreviation | Definition |
|---|---|
| Allele | One of two or more alternative forms of a gene at a given locus |
| Locus | Specific physical position of a gene on a chromosome |
| Genotype | The genetic constitution of an individual at one or more loci |
| Phenotype | The observable characteristics resulting from genotype + environment |
| Homozygous | Two identical alleles at a locus |
| Heterozygous | Two different alleles at a locus |
| Hemizygous | Only one allele present (e.g., X-linked genes in males) |
| Penetrance | Proportion of individuals with a genotype who show the phenotype |
| Expressivity | Degree to which a phenotype is expressed in an individual |
| Pleiotropy | One gene affecting multiple phenotypic traits |
| Epistasis | One gene masking or modifying expression of another gene |
| AD / AR | Autosomal dominant / autosomal recessive |
| XLR / XLD | X-linked recessive / X-linked dominant |
| LOH | Loss of heterozygosity |
| SNP | Single nucleotide polymorphism |
| CNV | Copy number variation |
| PCR | Polymerase chain reaction |
| FISH | Fluorescence in situ hybridization |
| NGS | Next-generation sequencing |
| CGH | Comparative genomic hybridization |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
Essential Genetic Quantities
| Parameter | Value |
|---|---|
| Human genome size | ~3.2 billion base pairs |
| Protein-coding genes | ~20,000–25,000 |
| Coding DNA (%) | ~1.5% of genome |
| Chromosome number | 46 (23 pairs: 22 autosomal + 1 sex pair) |
| Mitochondrial genome | 16,569 bp; 37 genes; circular, double-stranded |
03 DNA Structure, Chromatin & Chromosomes
Deoxyribonucleic acid (DNA) is the hereditary material of all cellular organisms. Its structure determines how genetic information is stored, replicated, and expressed.
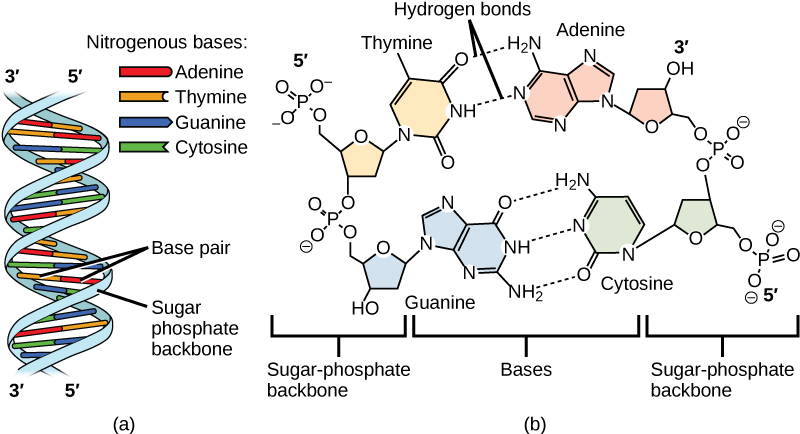
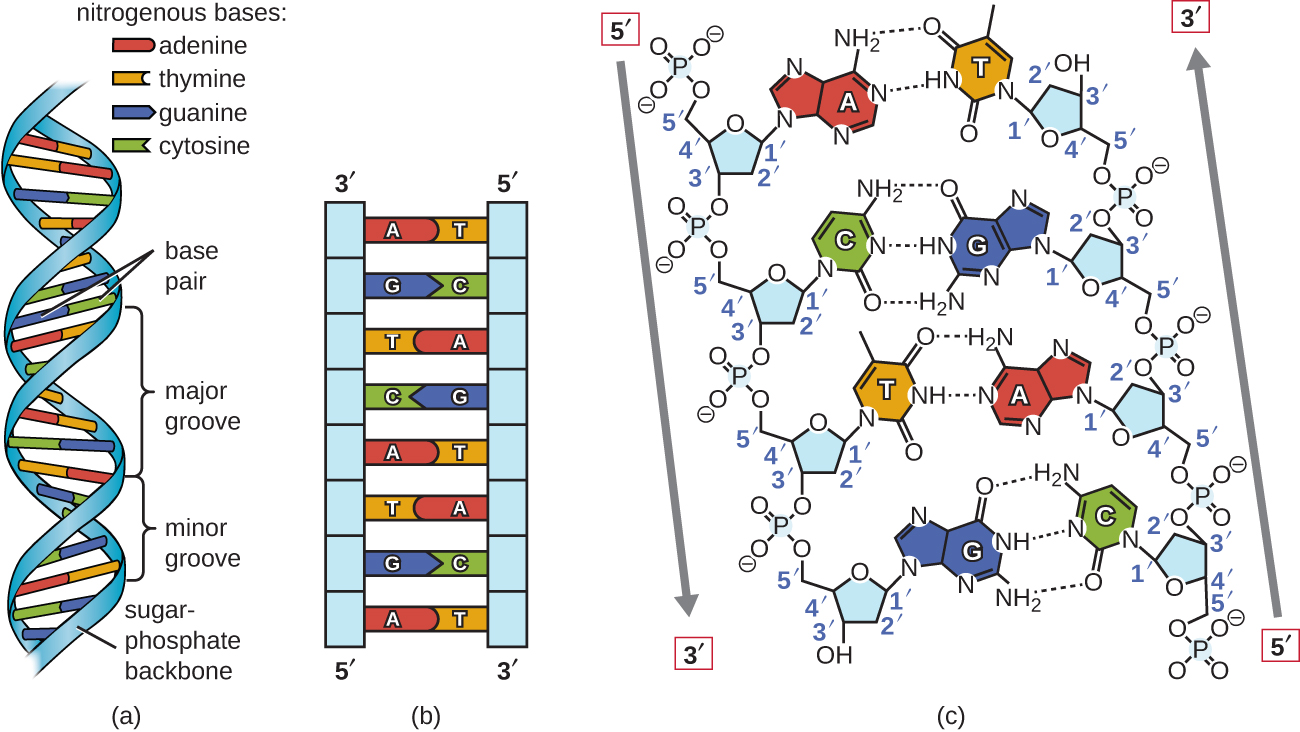
DNA Double Helix
- Sugar-phosphate backbone — alternating deoxyribose sugars and phosphodiester bonds; 5′ to 3′ directionality
- Base pairing — adenine (A) pairs with thymine (T) via 2 hydrogen bonds; guanine (G) pairs with cytosine (C) via 3 hydrogen bonds
- Antiparallel strands — the two strands run in opposite directions (5′→3′ and 3′→5′)
- Major and minor grooves — transcription factors and regulatory proteins bind primarily in the major groove
- B-form DNA — the predominant right-handed helix with ~10.5 bp per turn
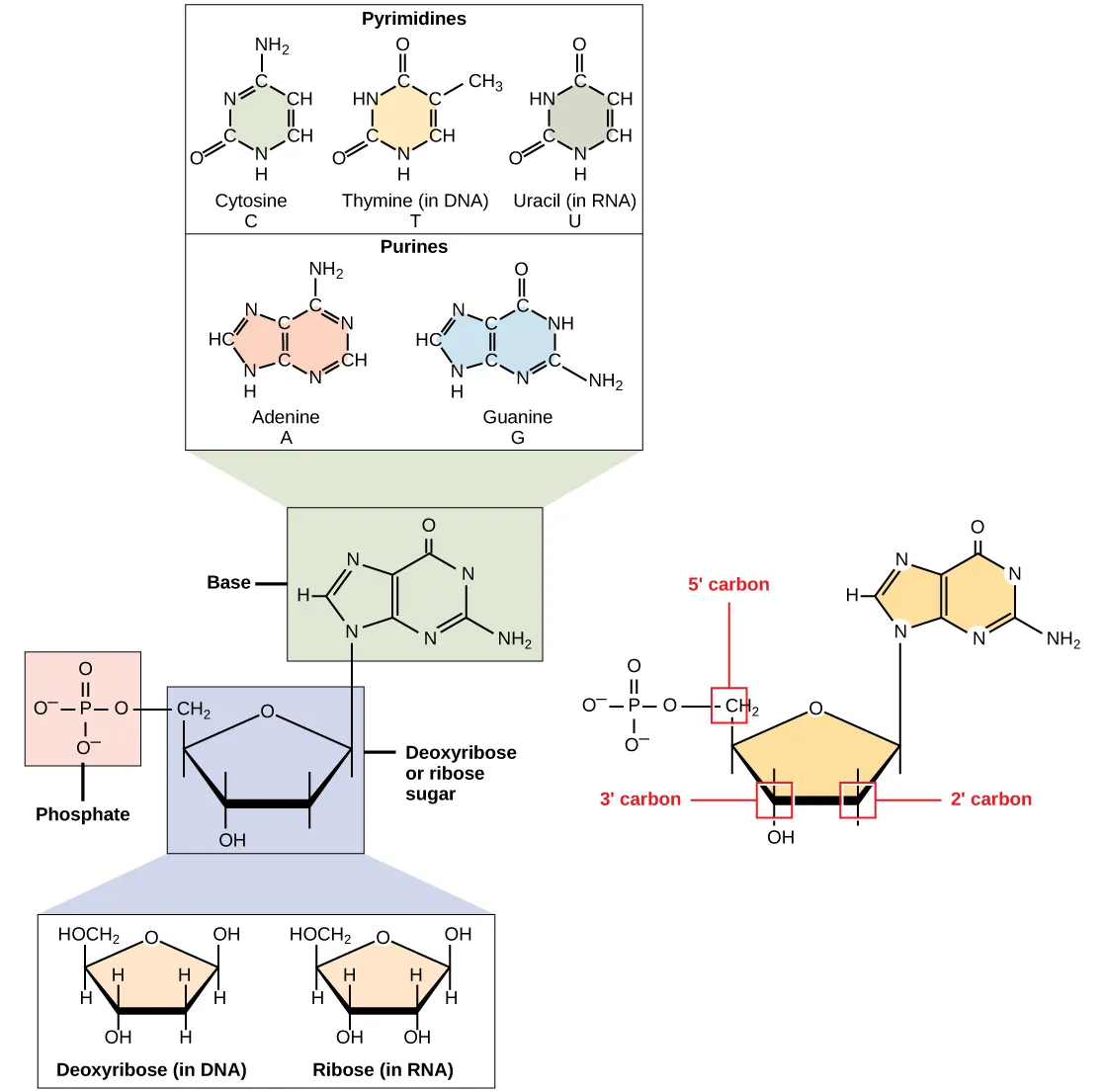
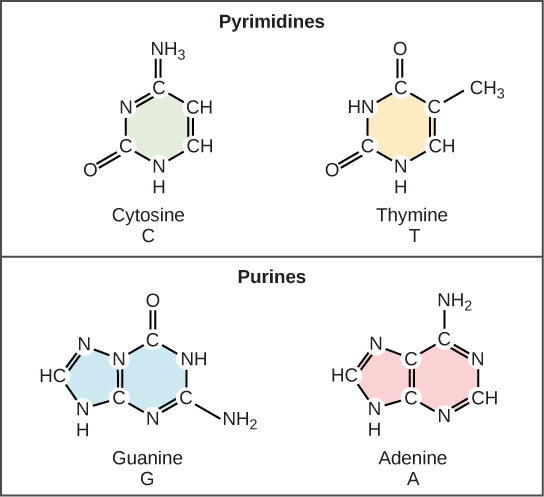
Nucleotide Chemistry
| Component | Purines (2 rings) | Pyrimidines (1 ring) |
|---|---|---|
| DNA bases | Adenine (A), Guanine (G) | Cytosine (C), Thymine (T) |
| RNA bases | Adenine (A), Guanine (G) | Cytosine (C), Uracil (U) |
| Mnemonic | PURe As Gold (purines: A, G) | CUT the PY (pyrimidines: C, U, T) |
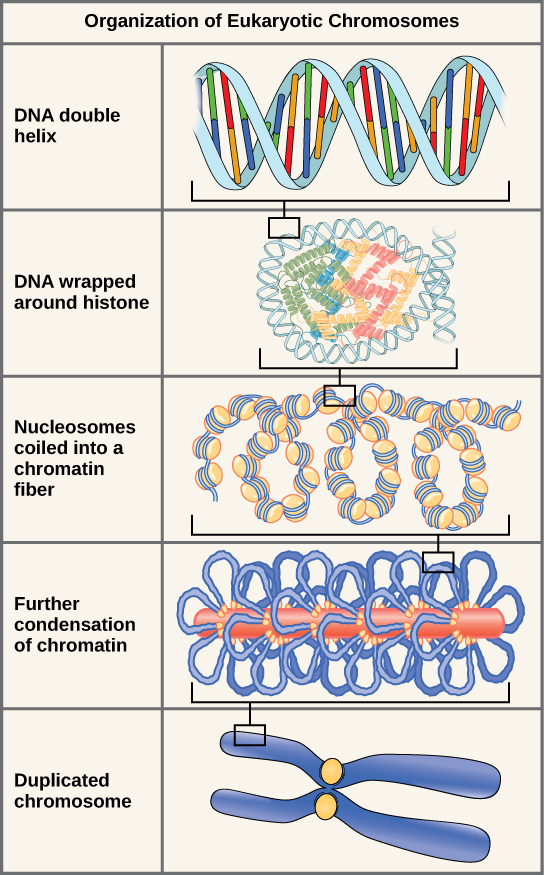
Chromatin Organization
| Level | Structure | Key Details |
|---|---|---|
| Nucleosome | DNA wrapped around histone octamer | ~147 bp of DNA wraps 1.65 turns around 2 copies each of H2A, H2B, H3, H4; linker histone H1 stabilizes |
| 30 nm fiber | Nucleosomes coiled into solenoid | Requires H1; compaction ~40-fold |
| Chromatin loops | Loops anchored to scaffold proteins | ~300 nm fiber; cohesin and condensin complexes |
| Metaphase chromosome | Maximum compaction | ~10,000-fold compaction; visible under light microscopy |
Euchromatin: loosely packed, transcriptionally active, appears light on staining. Heterochromatin: tightly packed, transcriptionally silent, appears dark. Constitutive heterochromatin is always condensed (centromeres, telomeres). Facultative heterochromatin is conditionally silenced (e.g., the inactive X chromosome — Barr body).
Histone Modifications
| Modification | Effect on Transcription | Mechanism |
|---|---|---|
| Acetylation (HATs) | Activates (opens chromatin) | Neutralizes positive charge on lysine residues → loosens DNA-histone interaction |
| Deacetylation (HDACs) | Represses (closes chromatin) | Restores positive charge → tighter DNA-histone binding |
| Methylation | Activates or represses | H3K4me3 = active; H3K9me3 and H3K27me3 = repressive |
| Phosphorylation | Activates (chromosome condensation) | H3S10 phosphorylation during mitosis |
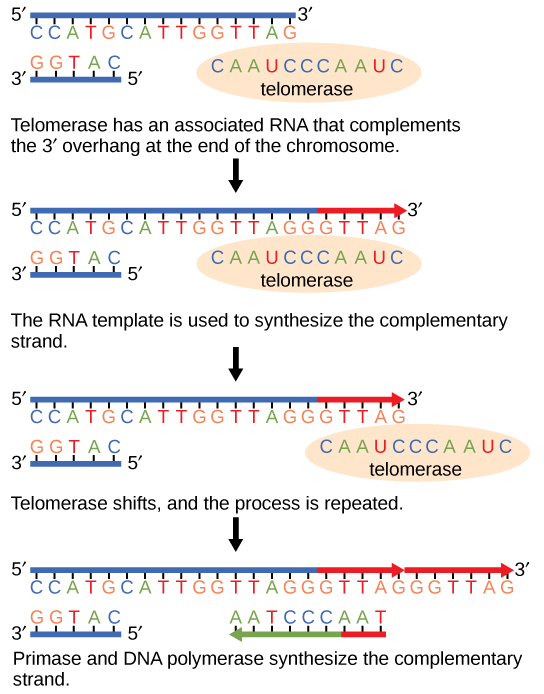
Telomeres
Telomeres are repetitive TTAGGG sequences at chromosome ends that protect against degradation, end-to-end fusion, and replication-associated shortening. Telomerase (a reverse transcriptase with an RNA template component, hTERC) adds telomeric repeats and is active in germ cells, stem cells, and ~90% of cancers. Somatic cells lack significant telomerase activity, leading to progressive telomere shortening with each division (end-replication problem) and eventual replicative senescence.
04 DNA Replication
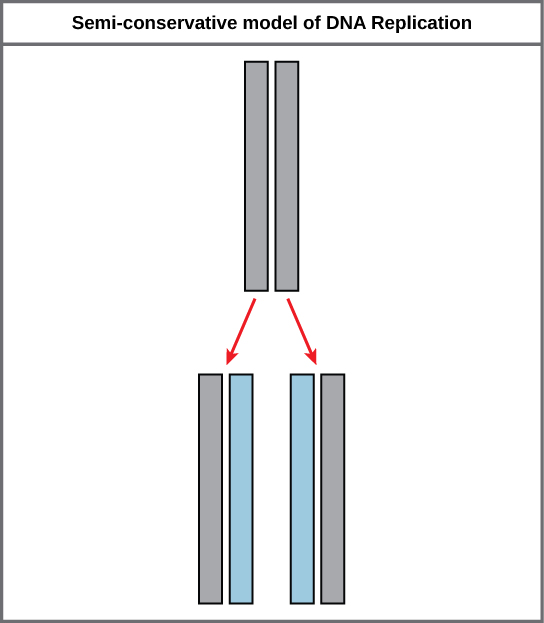
DNA replication is semiconservative (each daughter strand retains one parental strand), as demonstrated by the Meselson–Stahl experiment. Replication proceeds bidirectionally from origins of replication and requires a complex machinery of enzymes.
Replication Machinery
| Enzyme / Factor | Function | Clinical Relevance |
|---|---|---|
| Helicase | Unwinds dsDNA at the replication fork | ATP-dependent; creates single-stranded template |
| Single-strand binding proteins (SSBs) | Stabilize unwound ssDNA | Prevent reannealing and nuclease degradation |
| Topoisomerase I | Relaxes supercoils by creating single-strand nicks | Target of irinotecan and topotecan (camptothecins) |
| Topoisomerase II | Relieves supercoils by creating double-strand breaks | Target of etoposide and fluoroquinolones (bacterial) |
| Primase | Synthesizes RNA primer (~10 nt) | Required to initiate both leading and lagging strand synthesis |
| DNA Polymerase III (prokaryotic) | Main replicative polymerase; 5′→3′ synthesis + 3′→5′ proofreading | Eukaryotic equivalents: Pol ε (leading), Pol δ (lagging) |
| DNA Polymerase I (prokaryotic) | Removes RNA primers (5′→3′ exonuclease) and fills gaps | Also has 3′→5′ proofreading ability |
| DNA Ligase | Seals nicks between Okazaki fragments | Joins phosphodiester bonds; also used in DNA repair |
| Telomerase | Extends telomeres using RNA template | Reverse transcriptase; active in cancer cells |
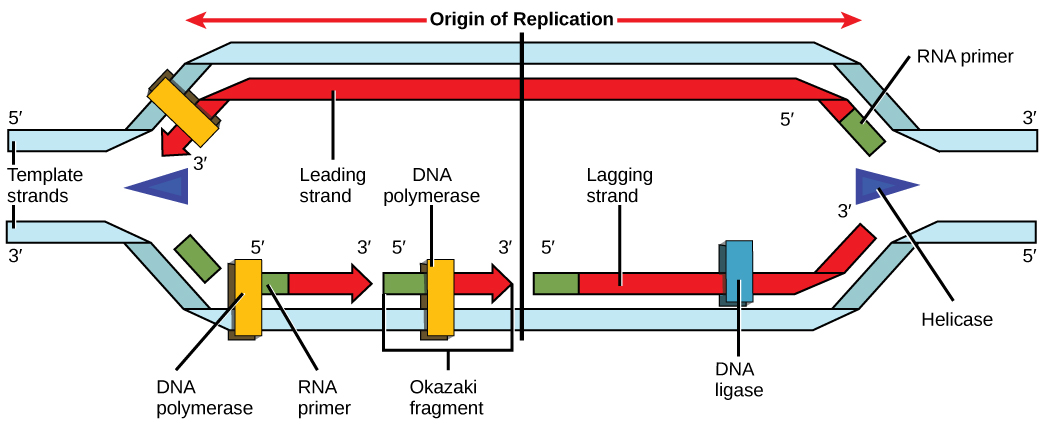
The leading strand is synthesized continuously in the 5′→3′ direction toward the replication fork. The lagging strand is synthesized discontinuously as Okazaki fragments (100–200 nt in eukaryotes) away from the fork, each requiring a separate RNA primer. DNA ligase seals the fragments together.
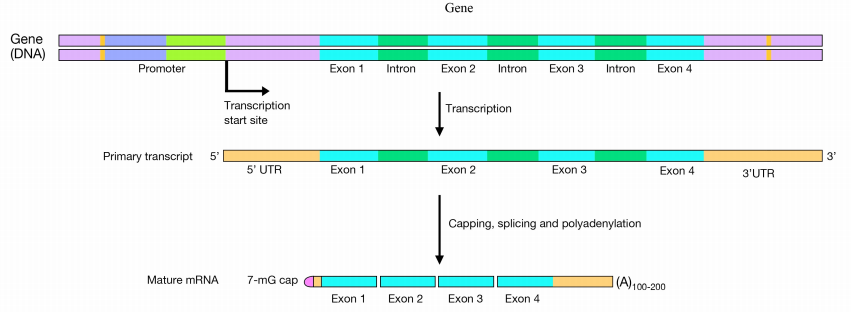
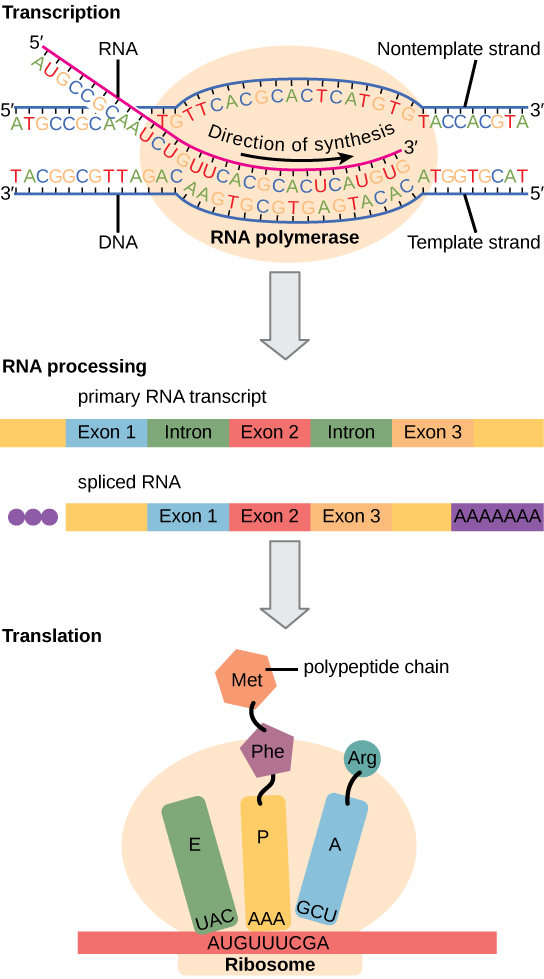
05 Transcription & RNA Processing
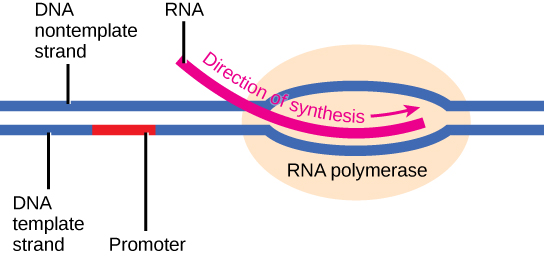
Transcription is the synthesis of RNA from a DNA template by RNA polymerase. In eukaryotes, three RNA polymerases exist, each responsible for different RNA species.
Eukaryotic RNA Polymerases
| Polymerase | Product | Location | Inhibitor |
|---|---|---|---|
| RNA Pol I | rRNA (28S, 18S, 5.8S) | Nucleolus | Not specifically targeted clinically |
| RNA Pol II | mRNA, snRNA, miRNA | Nucleoplasm | α-amanitin (mushroom toxin, Amanita phalloides) |
| RNA Pol III | tRNA, 5S rRNA | Nucleoplasm | α-amanitin at high doses |
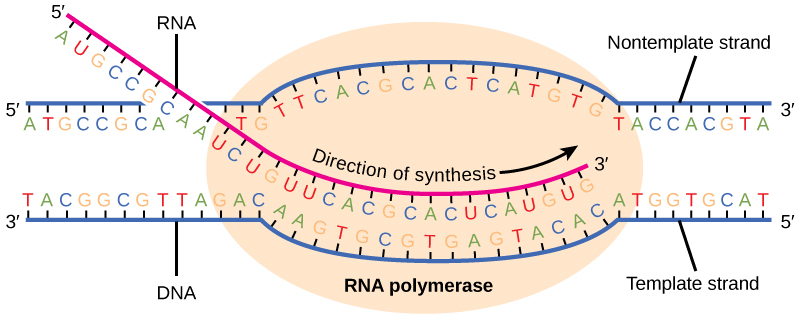
Steps of Eukaryotic Transcription
- Initiation — transcription factors (TFIID with TBP subunit) bind the TATA box (~25 bp upstream of start site) → recruit RNA Pol II → form pre-initiation complex
- Elongation — RNA Pol II synthesizes mRNA in the 5′→3′ direction, reading the template (antisense) strand 3′→5′
- Termination — polyadenylation signal (AAUAAA) triggers cleavage and release of transcript
Post-Transcriptional RNA Processing
| Modification | Description | Function |
|---|---|---|
| 5′ cap | 7-methylguanosine added to 5′ end | Protects from exonucleases; required for ribosome recognition and translation initiation |
| 3′ poly-A tail | ~200 adenine residues added by poly-A polymerase | Protects from degradation; facilitates nuclear export; aids translation |
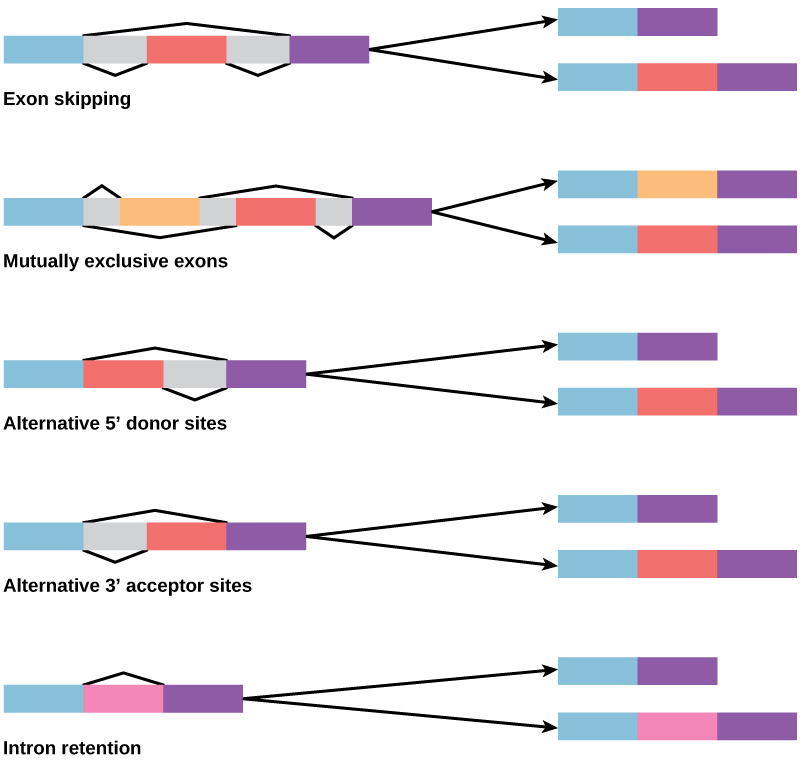
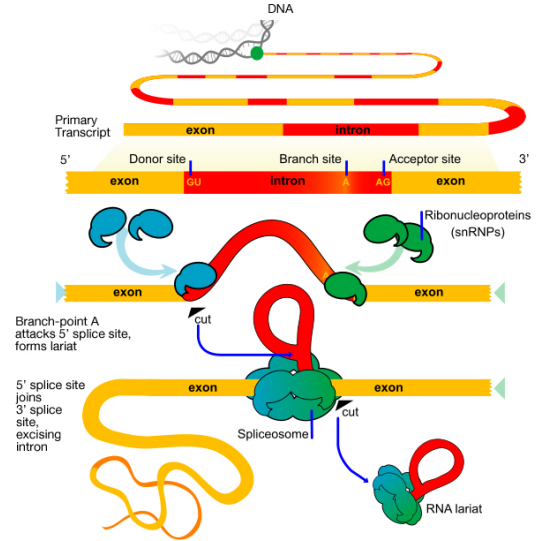
| Splicing | Introns removed, exons joined by spliceosome (snRNPs) | Allows alternative splicing → one gene can produce multiple proteins |
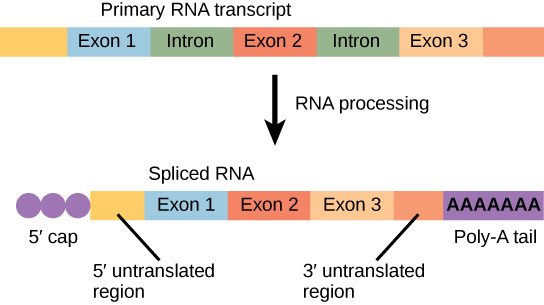
Introns begin with GU (donor site) and end with AG (acceptor site) — the “GU-AG rule.” The spliceosome contains snRNPs (small nuclear ribonucleoproteins, pronounced “snurps”) made of snRNA + protein. Anti-snRNP antibodies (anti-Smith, anti-U1 RNP) are seen in SLE and mixed connective tissue disease, respectively.
Types of RNA
| RNA Type | Function | Key Details |
|---|---|---|
| mRNA | Carries genetic code from DNA to ribosomes | Made by RNA Pol II; processed (cap, tail, splicing); codon sequence |
| tRNA | Adaptor molecule; carries amino acids to ribosome | Cloverleaf structure; anticodon loop; 3′ CCA acceptor stem; charged by aminoacyl-tRNA synthetase |
| rRNA | Structural and catalytic component of ribosomes | 28S, 18S, 5.8S (Pol I); 5S (Pol III); peptidyl transferase is a ribozyme (23S rRNA) |
| snRNA | Component of spliceosome (snRNPs) | U1, U2, U4, U5, U6; anti-Smith Ab targets snRNPs in SLE |
| miRNA | Post-transcriptional gene silencing | ~22 nt; binds 3′ UTR; RISC complex; widespread role in development and cancer |
| siRNA | Targeted mRNA degradation | Double-stranded; therapeutic applications (patisiran for TTR amyloidosis) |
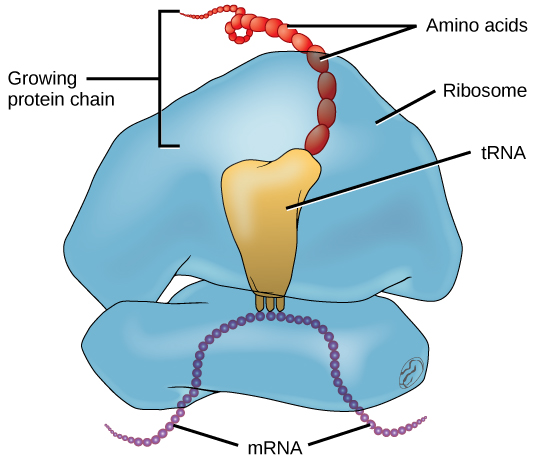
06 Translation & the Genetic Code
Translation is the process by which ribosomes decode mRNA into polypeptide chains. It occurs in the cytoplasm (free ribosomes) or on the rough endoplasmic reticulum (secreted and membrane proteins).
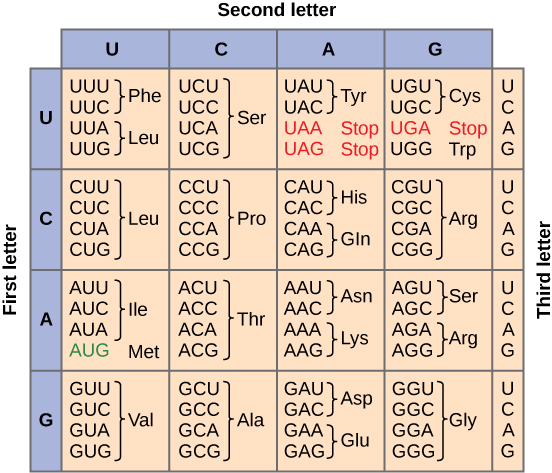
The Genetic Code
- Triplet — each codon consists of 3 nucleotides encoding one amino acid
- Degenerate (redundant) — most amino acids are specified by more than one codon (wobble position = 3rd base)
- Non-overlapping — codons are read sequentially without overlap
- Universal — the same code is used by virtually all organisms (minor exceptions in mitochondria)
- Unambiguous — each codon specifies only one amino acid
- Start codon — AUG (methionine); also serves as internal methionine codon
- Stop codons — UAA, UAG, UGA (“U Are Annoying, U Are Gone, U Go Away”)
Steps of Translation
| Phase | Events | Key Factors |
|---|---|---|
| Initiation | Small ribosomal subunit (40S) binds 5′ cap, scans for AUG start codon; large subunit (60S) joins | eIF (eukaryotic initiation factors); initiator Met-tRNA binds P site |
| Elongation | Aminoacyl-tRNA enters A site → peptide bond formed (peptidyl transferase, a ribozyme) → ribosome translocates | EF-Tu (prokaryotic) / eEF-1 (eukaryotic); GTP hydrolysis; A site → P site → E site |
| Termination | Stop codon in A site → release factors bind → polypeptide released | RF1, RF2, RF3 (prokaryotic); eRF1, eRF3 (eukaryotic) |
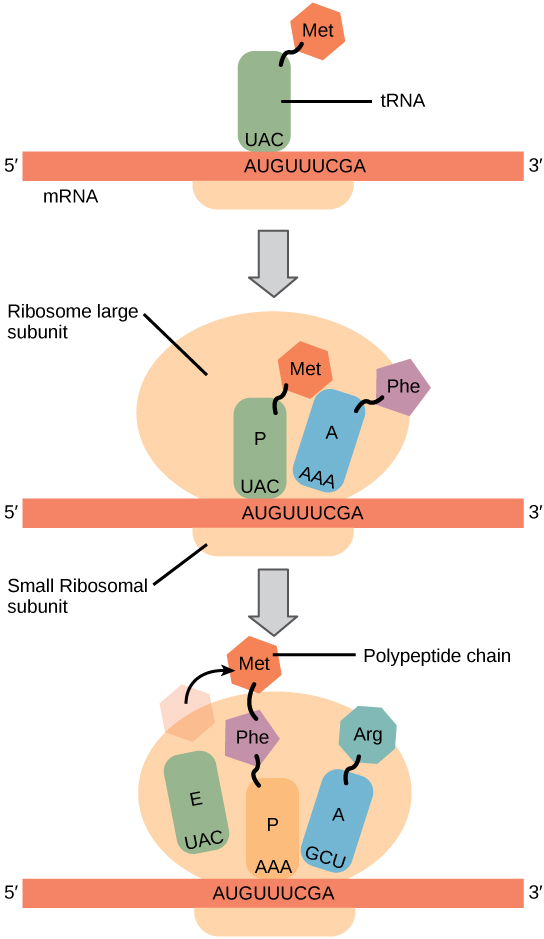
Antibiotics Targeting Translation
| Antibiotic | Target | Mechanism |
|---|---|---|
| Aminoglycosides (gentamicin) | 30S subunit | Cause mRNA misreading; bactericidal |
| Tetracyclines | 30S subunit | Block aminoacyl-tRNA binding to A site; bacteriostatic |
| Chloramphenicol | 50S subunit | Inhibits peptidyl transferase; bacteriostatic |
| Macrolides (erythromycin) | 50S subunit | Block translocation; bacteriostatic |
| Clindamycin | 50S subunit | Block translocation; bacteriostatic |
| Linezolid | 50S subunit | Prevents 70S initiation complex formation |
Post-Translational Modifications
- Signal peptide cleavage — signal recognition particle (SRP) directs nascent proteins to ER
- Glycosylation — N-linked (asparagine, in ER) and O-linked (serine/threonine, in Golgi)
- Phosphorylation — by kinases on Ser, Thr, Tyr residues; key signaling mechanism
- Ubiquitination — tags proteins for proteasomal degradation
- Hydroxylation — proline and lysine hydroxylation in collagen (requires vitamin C)
Protein Trafficking Destinations
| Destination | Signal / Mechanism | Examples |
|---|---|---|
| Rough ER | N-terminal signal peptide recognized by SRP | Secreted proteins (insulin, antibodies), membrane proteins |
| Lysosomes | Mannose-6-phosphate tag added in cis-Golgi | Acid hydrolases (hexosaminidase, glucocerebrosidase) |
| Nucleus | Nuclear localization signal (NLS); importins | Transcription factors, histones |
| Mitochondria | N-terminal amphipathic helix; TOM/TIM complexes | Mitochondrial matrix enzymes (most encoded by nuclear DNA) |
| Peroxisomes | PTS1 (C-terminal SKL) or PTS2 signal | Catalase, β-oxidation of very long chain fatty acids |
| Proteasome | Polyubiquitin tag (K48-linked) | Misfolded proteins, cell cycle regulators (cyclins) |
Collagen synthesis is a high-yield topic that integrates genetics, biochemistry, and clinical medicine. Key steps: (1) Translation of preprocollagen → (2) Hydroxylation of proline and lysine (requires vitamin C) in ER → (3) Glycosylation in ER → (4) Triple helix formation (procollagen) → (5) Secretion → (6) Cleavage of propeptides to tropocollagen → (7) Cross-linking by lysyl oxidase (requires copper). Defects: Osteogenesis imperfecta (type I collagen), Ehlers–Danlos (various collagen types), Scurvy (vitamin C deficiency → defective hydroxylation), Menkes disease (copper deficiency → defective cross-linking).
07 Regulation of Gene Expression
Gene expression is regulated at multiple levels — from chromatin remodeling to post-translational modification. Understanding these mechanisms is critical for comprehending disease pathogenesis and therapeutic targets.
Levels of Gene Regulation
| Level | Mechanism | Examples |
|---|---|---|
| Epigenetic | DNA methylation (CpG islands); histone modification | Imprinting; X-inactivation; cancer epigenetics |
| Transcriptional | Transcription factors, enhancers, silencers, promoters | p53 activates p21 transcription; steroid hormone receptors |
| Post-transcriptional | mRNA stability, alternative splicing, RNA interference (miRNA, siRNA) | Iron response elements control ferritin/transferrin receptor mRNA |
| Translational | Regulation of ribosome binding and scanning | mTOR pathway; eIF4E regulation |
| Post-translational | Protein modification, folding, degradation | Ubiquitin-proteasome pathway; phosphorylation cascades |
DNA methyltransferases add methyl groups to cytosine at CpG dinucleotides. Methylation of CpG islands in promoter regions generally silences gene expression. Hypermethylation of tumor suppressor gene promoters is a common mechanism of cancer development (e.g., hypermethylation of MLH1 in sporadic microsatellite-unstable colon cancer). Conversely, global hypomethylation can activate oncogenes.
Regulatory RNA Species
- miRNA — small (~22 nt) non-coding RNA; binds 3′ UTR of target mRNA → translational repression or mRNA degradation; Drosha processes in nucleus, Dicer processes in cytoplasm
- siRNA — small interfering RNA; double-stranded; induces RISC-mediated mRNA cleavage (used therapeutically, e.g., patisiran for TTR amyloidosis, inclisiran for PCSK9)
- lncRNA — long non-coding RNA (>200 nt); roles in X-inactivation (XIST RNA coats inactive X), imprinting, chromatin remodeling
- Antisense oligonucleotides (ASOs) — synthetic single-stranded nucleic acids that bind target mRNA; modulate splicing or block translation (nusinersen for SMA, mipomersen for familial hypercholesterolemia)
- Ribozymes — catalytic RNA molecules; peptidyl transferase (23S rRNA) is the classic example; self-splicing introns (Group I and II introns)
Cell Signaling Pathways Relevant to Gene Regulation
| Pathway | Key Components | Function | Clinical Relevance |
|---|---|---|---|
| RAS-MAPK | RAS → RAF → MEK → ERK | Cell proliferation, differentiation | RAS mutations in ~30% of cancers; BRAF V600E in melanoma (vemurafenib); RASopathies (Noonan, Costello syndromes) |
| JAK-STAT | Cytokine receptor → JAK → STAT | Hematopoiesis, immune function | JAK2 V617F in polycythemia vera, essential thrombocythemia; ruxolitinib (JAK inhibitor) |
| PI3K-AKT-mTOR | PI3K → PIP3 → AKT → mTOR | Cell survival, growth, metabolism | PTEN (negative regulator) mutations in Cowden syndrome; everolimus for tuberous sclerosis |
| Wnt/β-catenin | Wnt → Frizzled → β-catenin stabilization | Cell proliferation, stem cell maintenance | APC loss → constitutive Wnt signaling → FAP and colorectal cancer |
| Notch | Notch receptor → intracellular domain → transcription | Cell fate determination | Gain-of-function Notch1 mutations in T-ALL; Alagille syndrome (JAG1/NOTCH2 loss) |
| Hedgehog | Shh → Patched → Smoothened → Gli | Embryonic development, cell proliferation | PTCH1 mutations in Gorlin syndrome (basal cell nevus syndrome); vismodegib inhibits Smoothened |
08 Types of Mutations
A mutation is any heritable change in the DNA sequence. Mutations range from single nucleotide changes to large chromosomal rearrangements and are the fundamental basis of genetic disease, evolution, and cancer.
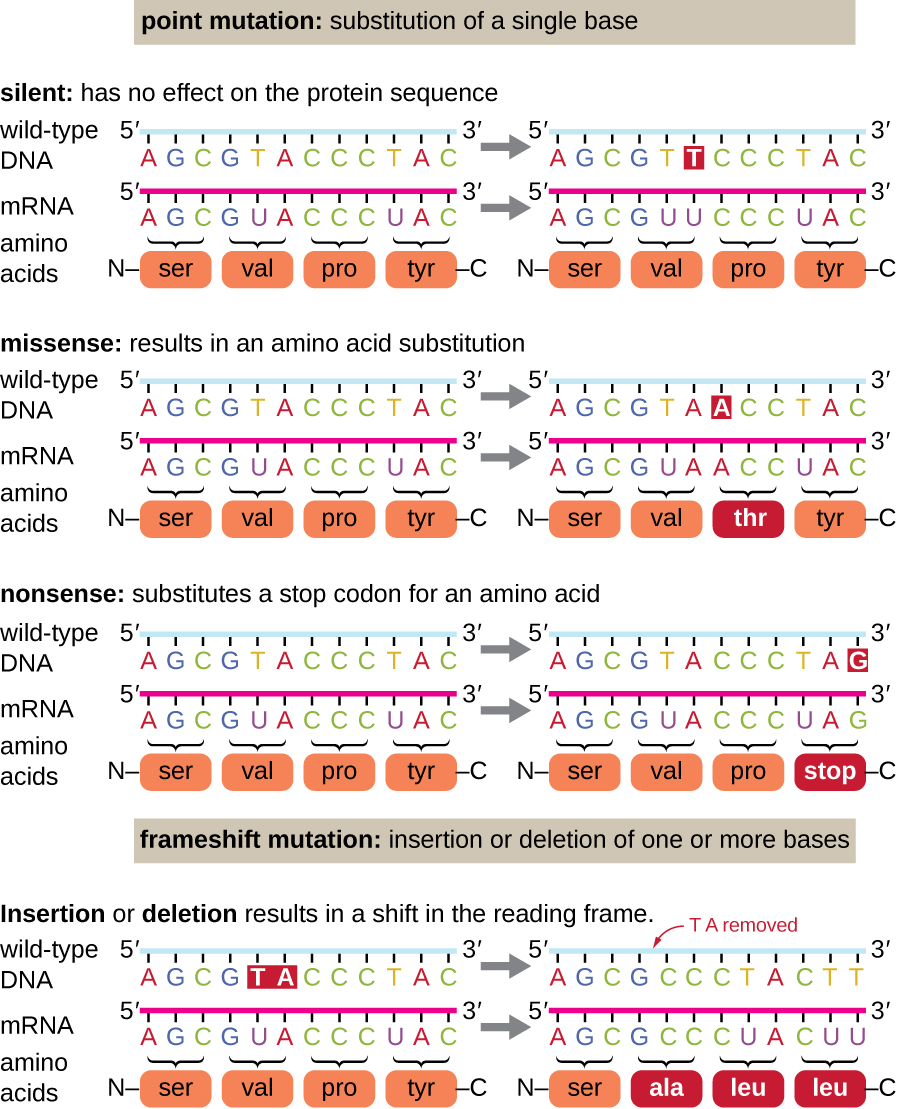
Point Mutations (Single Nucleotide)
| Type | Definition | Classic Example |
|---|---|---|
| Silent (synonymous) | Nucleotide change that does not alter the amino acid (due to codon degeneracy) | Often at wobble (3rd) position |
| Missense | Nucleotide change that results in a different amino acid | Sickle cell disease: Glu → Val at position 6 of β-globin (GAG → GTG) |
| Nonsense | Nucleotide change that creates a premature stop codon | Some forms of β-thalassemia; Duchenne muscular dystrophy |
Missense Mutation Subtypes
- Conservative — substituted amino acid has similar properties (e.g., Asp → Glu, both acidic); often tolerated
- Non-conservative — substituted amino acid has different properties (e.g., Glu → Val in sickle cell); more likely pathogenic
Frameshift Mutations
| Type | Mechanism | Result |
|---|---|---|
| Insertion | Addition of nucleotides (not multiples of 3) | Alters reading frame downstream → garbled protein |
| Deletion | Loss of nucleotides (not multiples of 3) | Alters reading frame downstream → garbled protein |
Duchenne: frameshift deletion in dystrophin gene → premature stop → no functional dystrophin → severe, early-onset. Becker: in-frame deletion → truncated but partially functional dystrophin → milder, later onset. This exemplifies how the same gene can produce different severity depending on whether the reading frame is preserved.
Splice Site Mutations
Mutations at intron-exon boundaries (GT donor or AG acceptor sites) cause exon skipping, intron retention, or activation of cryptic splice sites. Many cases of β-thalassemia result from splice site mutations.
Other Mutation Types
- Trinucleotide repeat expansion — unstable repeats that grow with each generation (see Section 14)
- Loss-of-function — reduced or absent protein function; typically recessive
- Gain-of-function — new or enhanced protein activity; typically dominant
- Dominant-negative — mutant protein interferes with wild-type protein function (e.g., mutant p53, osteogenesis imperfecta type II)
Transition vs. Transversion
- Transition — purine → purine (A↔G) or pyrimidine → pyrimidine (C↔T); more common
- Transversion — purine ↔ pyrimidine; less common but more likely to be non-conservative
09 DNA Damage & Repair Mechanisms
DNA is continuously damaged by endogenous (replication errors, oxidative stress) and exogenous (UV, radiation, chemicals) agents. Multiple repair pathways exist to maintain genomic integrity, and defects in these pathways are the basis of many cancer predisposition syndromes.
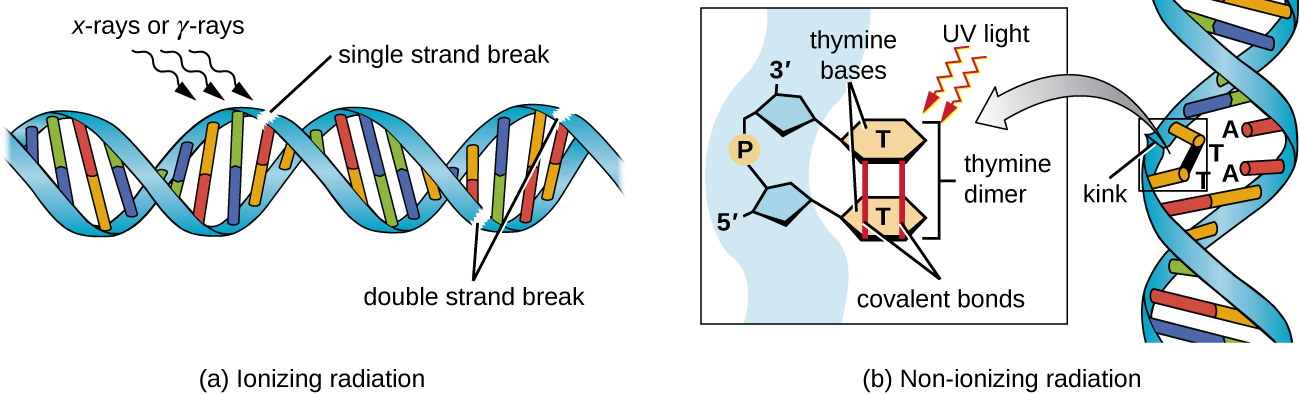
DNA Repair Pathways
| Pathway | Damage Repaired | Mechanism | Defect Causes |
|---|---|---|---|
| Nucleotide Excision Repair (NER) | Bulky adducts, thymine dimers (UV damage) | Endonucleases excise ~30 nt segment; DNA Pol fills gap; ligase seals | Xeroderma pigmentosum — extreme UV sensitivity, >1000× skin cancer risk |
| Base Excision Repair (BER) | Small base modifications (deamination, oxidation, alkylation) | Glycosylase removes damaged base → AP endonuclease → Pol β fills → ligase seals | Defects associated with cancer susceptibility |
| Mismatch Repair (MMR) | Base-base mismatches, insertion/deletion loops from replication errors | MSH2/MLH1 recognize mismatch → excise segment → resynthesize | Lynch syndrome (HNPCC) — hereditary nonpolyposis colorectal cancer |
| Homologous Recombination (HR) | Double-strand breaks (DSBs) | Uses sister chromatid as template; error-free; BRCA1/BRCA2 essential | BRCA mutations — breast, ovarian, prostate cancer |
| Non-Homologous End Joining (NHEJ) | Double-strand breaks | Directly ligates broken ends; error-prone (may lose nucleotides) | Defects in Ku70/Ku80; associated with immunodeficiency and cancer |
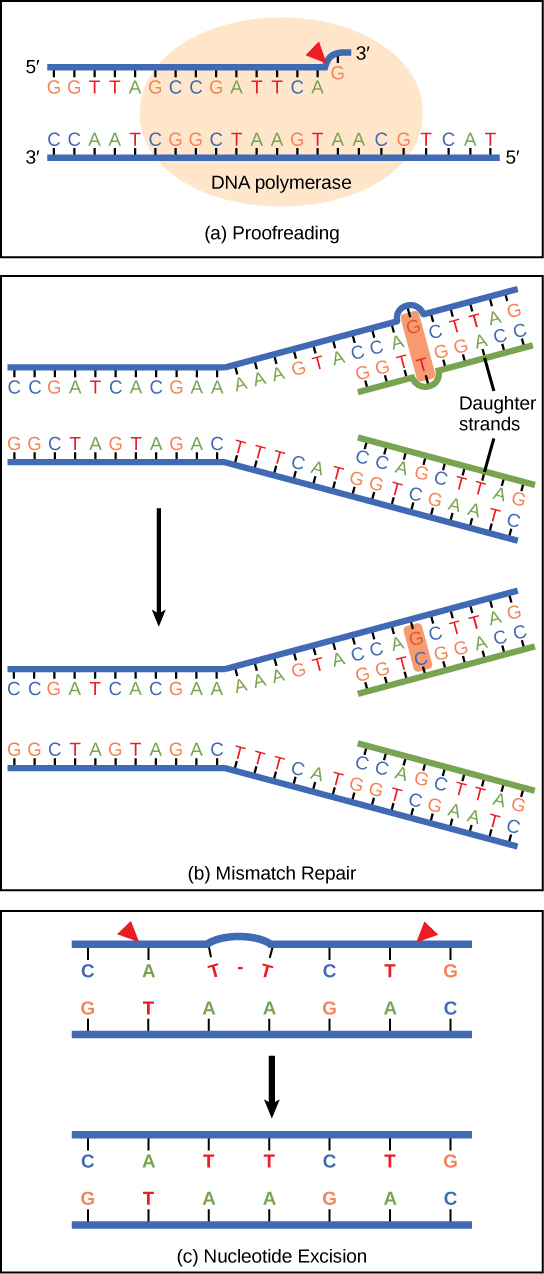
XP is an autosomal recessive disorder with defective nucleotide excision repair. Patients cannot repair UV-induced thymine dimers and have dramatically increased risk of skin cancers (basal cell, squamous cell, melanoma) in sun-exposed areas, often presenting in childhood. They must strictly avoid sun exposure.
Common DNA Damaging Agents
| Agent | Type of Damage | Repair Pathway |
|---|---|---|
| UV light (UVB) | Thymine (pyrimidine) dimers | NER |
| Ionizing radiation | Double-strand breaks, base modifications | HR, NHEJ |
| Alkylating agents (cyclophosphamide) | Alkylated bases, cross-links | BER, NER |
| Reactive oxygen species | 8-oxoguanine, strand breaks | BER |
| Spontaneous deamination | Cytosine → uracil; 5-methylcytosine → thymine | BER (uracil-DNA glycosylase) |
Cancer-Associated DNA Repair Syndromes Summary
| Syndrome | Defective Pathway | Inheritance | Cancer Risk | Other Features |
|---|---|---|---|---|
| Xeroderma pigmentosum | NER | AR | Skin cancers (>1000×) | Extreme photosensitivity; neurodegeneration in some subtypes |
| Lynch syndrome | MMR | AD | CRC, endometrial, ovarian | Microsatellite instability; right-sided colon cancers |
| BRCA1/2 | HR | AD | Breast, ovarian, prostate, pancreatic | PARP inhibitor sensitivity (synthetic lethality) |
| Fanconi anemia | Cross-link repair | AR | AML, squamous cell cancers | Bone marrow failure, short stature, radial defects |
| Ataxia-telangiectasia | DSB signaling (ATM) | AR | Lymphoma, leukemia | Cerebellar ataxia, telangiectasias, IgA deficiency, radiosensitivity |
| Bloom syndrome | RecQ helicase (BLM) | AR | Multiple cancer types | Growth restriction, sun-sensitive facial rash, sister chromatid exchanges |
10 Autosomal Dominant Inheritance
Autosomal dominant (AD) disorders require only one mutant allele for phenotypic expression. Affected individuals are typically heterozygous (Aa). Each child of an affected parent has a 50% chance of inheriting the disorder.
Characteristics of AD Inheritance
- Vertical transmission (affected individuals in every generation)
- Males and females equally affected
- Male-to-male transmission possible (distinguishes from X-linked)
- Unaffected individuals do not transmit the trait
- Variable expressivity and incomplete penetrance are common
- Often involves structural proteins or regulatory proteins
High-Yield AD Disorders
| Disorder | Gene / Protein | Key Features |
|---|---|---|
| Marfan syndrome | FBN1 (fibrillin-1) | Tall stature, arachnodactyly, lens subluxation (up and out), aortic root dilation, MVP |
| Ehlers–Danlos (classic) | COL5A1/2 (type V collagen) | Skin hyperextensibility, joint hypermobility, easy bruising |
| Osteogenesis imperfecta (type I) | COL1A1/2 (type I collagen) | Brittle bones, blue sclerae, hearing loss, dental abnormalities |
| Huntington disease | HTT (huntingtin) | CAG repeat expansion; chorea, dementia, psychiatric symptoms; onset ~40 years |
| Familial hypercholesterolemia | LDLR | Elevated LDL, xanthomas, premature atherosclerosis |
| Hereditary spherocytosis | Ankyrin, spectrin, band 3 | Spherocytes, hemolytic anemia, splenomegaly, positive osmotic fragility test |
| von Willebrand disease (type 1) | VWF | Most common inherited bleeding disorder; mucocutaneous bleeding; prolonged bleeding time |
| Neurofibromatosis type 1 | NF1 (neurofibromin) | Café-au-lait spots, neurofibromas, Lisch nodules, optic gliomas |
| Tuberous sclerosis | TSC1/TSC2 (hamartin/tuberin) | Cortical tubers, subependymal nodules, facial angiofibromas, cardiac rhabdomyomas, renal angiomyolipomas |
| ADPKD | PKD1 (85%) / PKD2 | Bilateral renal cysts, hepatic cysts, berry aneurysms, mitral valve prolapse |
Autosomal Dominant Connective Tissue Disorders Compared
| Feature | Marfan | Ehlers–Danlos (Classic) | Osteogenesis Imperfecta (Type I) |
|---|---|---|---|
| Gene | FBN1 (fibrillin-1) | COL5A1/2 | COL1A1/2 |
| Skeletal | Tall, arachnodactyly, pectus excavatum | Joint hypermobility | Fractures, short stature (in severe types) |
| Skin | Striae | Hyperextensible, easy bruising | Thin, translucent |
| Eyes | Lens subluxation (up and temporal) | Usually normal | Blue sclerae |
| Cardiovascular | Aortic root dilation, MVP, dissection | Vascular type: arterial/organ rupture (COL3A1) | Aortic root dilation (less common) |
| Key distinction | Homocystinuria mimics but has downward lens subluxation | Vascular type is life-threatening | Hearing loss (otosclerosis) |
11 Autosomal Recessive Inheritance
Autosomal recessive (AR) disorders require two mutant alleles (homozygous aa) for phenotypic expression. Heterozygous carriers (Aa) are typically unaffected. When two carriers mate, each child has a 25% chance of being affected, a 50% chance of being a carrier, and a 25% chance of being unaffected.
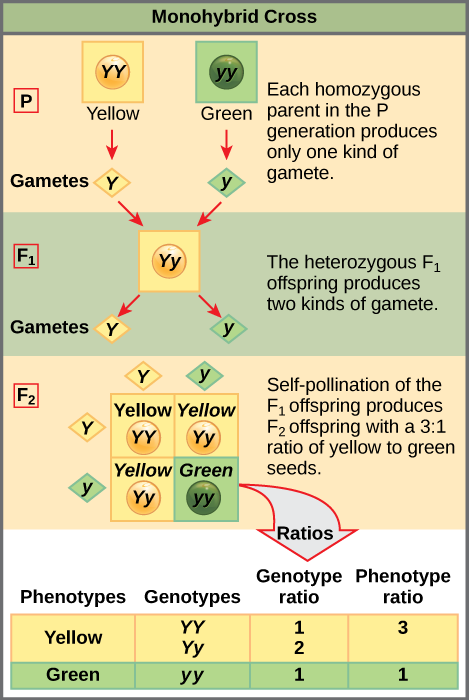
Characteristics of AR Inheritance
- Horizontal pattern (affected siblings, unaffected parents)
- Parents are typically heterozygous carriers
- Consanguinity increases risk
- Males and females equally affected
- Often involves enzyme deficiencies (inborn errors of metabolism)
- Carrier frequency can be calculated from disease prevalence using Hardy–Weinberg
High-Yield AR Disorders
| Disorder | Gene / Enzyme | Key Features |
|---|---|---|
| Cystic fibrosis | CFTR (ΔF508 most common) | Thick mucus, recurrent pulmonary infections, pancreatic insufficiency, meconium ileus, infertility (males) |
| Sickle cell disease | HBB (β-globin) | Vaso-occlusive crises, splenic sequestration, acute chest syndrome; Glu→Val at position 6 |
| Phenylketonuria (PKU) | PAH (phenylalanine hydroxylase) | Intellectual disability, musty body odor, fair skin/hair, eczema; treated with dietary Phe restriction |
| Tay–Sachs disease | HEXA (hexosaminidase A) | GM2 ganglioside accumulation; cherry-red macula, progressive neurodegeneration; Ashkenazi Jewish |
| Gaucher disease | GBA (glucocerebrosidase) | Glucocerebroside accumulation; hepatosplenomegaly, bone crises, Gaucher cells (crinkled paper macrophages) |
| Galactosemia | GALT (galactose-1-phosphate uridylyltransferase) | Jaundice, hepatomegaly, cataracts, E. coli sepsis in neonates |
| Wilson disease | ATP7B | Copper accumulation; hepatic disease, Kayser–Fleischer rings, neuropsychiatric symptoms |
| Hemochromatosis | HFE (C282Y) | Iron overload; cirrhosis, diabetes, cardiomyopathy, skin bronzing, arthropathy |
| α1-antitrypsin deficiency | SERPINA1 (PiZZ) | Panacinar emphysema (lower lobes), hepatic cirrhosis (PAS-positive globules) |
Lysosomal Storage Diseases (AR)
| Disease | Enzyme Deficiency | Accumulated Substrate | Key Features |
|---|---|---|---|
| Tay–Sachs | Hexosaminidase A | GM2 ganglioside | Cherry-red macula, progressive neurodegeneration, death by ~4 years; no hepatosplenomegaly |
| Niemann–Pick (A/B) | Sphingomyelinase | Sphingomyelin | Type A: neurodegeneration + hepatosplenomegaly + cherry-red macula; “foam cells” |
| Gaucher (type 1) | Glucocerebrosidase | Glucocerebroside | Hepatosplenomegaly, pancytopenia, bone crises; “crinkled paper” macrophages; enzyme replacement available |
| Krabbe | Galactosylceramidase | Galactocerebroside | Globoid cells; peripheral neuropathy, optic atrophy, developmental regression |
| Metachromatic leukodystrophy | Arylsulfatase A | Sulfatides | Central and peripheral demyelination; metachromatic granules |
| Hurler (MPS I) | α-L-iduronidase | Heparan/dermatan sulfate | Coarse facies, corneal clouding, hepatosplenomegaly, skeletal abnormalities; AR (not X-linked like Hunter) |
| Pompe | Acid maltase (α-glucosidase) | Glycogen (in lysosomes) | Infantile: hypertrophic cardiomyopathy, hypotonia, death <2 years; enzyme replacement available |
12 X-Linked & Mitochondrial Inheritance
X-Linked Recessive (XLR)
XLR disorders predominantly affect males (hemizygous, XY) because they have only one X chromosome. Carrier females (heterozygous) are usually unaffected but may show mild manifestations due to skewed X-inactivation.
- No male-to-male transmission (father passes Y to sons)
- Affected males transmit the carrier state to all daughters
- Carrier mothers have a 50% chance of affected sons and 50% carrier daughters
High-Yield XLR Disorders
| Disorder | Gene / Protein | Key Features |
|---|---|---|
| Duchenne muscular dystrophy | DMD (dystrophin) | Frameshift mutation; progressive proximal muscle weakness, calf pseudohypertrophy, Gowers sign; elevated CK; death by ~20s |
| Hemophilia A | F8 (factor VIII) | Hemarthroses, deep tissue bleeding; prolonged PTT, normal PT and bleeding time |
| Hemophilia B | F9 (factor IX) | Clinically identical to hemophilia A; “Christmas disease” |
| G6PD deficiency | G6PD | Episodic hemolytic anemia triggered by oxidative stress (fava beans, sulfonamides, primaquine); Heinz bodies, bite cells |
| Fabry disease | GLA (α-galactosidase A) | Ceramide trihexoside accumulation; angiokeratomas, acroparesthesias, renal failure, corneal dystrophy |
| Hunter syndrome (MPS II) | IDS (iduronate-2-sulfatase) | Like Hurler but milder; no corneal clouding (only MPS without corneal clouding); aggressive behavior |
| Lesch–Nyhan syndrome | HPRT1 | HGPRT deficiency; hyperuricemia, gout, intellectual disability, self-mutilation |
| Bruton agammaglobulinemia | BTK | Absent mature B cells; recurrent bacterial infections after 6 months (maternal IgG wanes) |
X-Linked Dominant (XLD)
Rare. Affected mothers transmit to 50% of sons and 50% of daughters. Affected fathers transmit to all daughters and no sons. Some XLD conditions are lethal in hemizygous males.
- Rett syndrome — MECP2 mutation; almost exclusively in girls (lethal in males); stereotypic hand-wringing, regression after 6–18 months
- Incontinentia pigmenti — IKBKG/NEMO; skin lesions in lines of Blaschko; lethal in males
- Alport syndrome (most common form) — COL4A5; sensorineural hearing loss, ocular abnormalities, progressive glomerulonephritis

Mitochondrial (Maternal) Inheritance
Mitochondrial DNA (mtDNA) is inherited exclusively from the mother. All children of an affected mother may be affected; an affected father cannot transmit the disease. Variable expression due to heteroplasmy (mixture of normal and mutant mitochondria).
| Disorder | Key Features |
|---|---|
| MELAS | Mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes |
| MERRF | Myoclonic epilepsy with ragged red fibers |
| Leber hereditary optic neuropathy | Bilateral painless vision loss in young adults; affects males more often |
| Kearns–Sayre syndrome | Progressive external ophthalmoplegia, pigmentary retinopathy, cardiac conduction defects; large mtDNA deletion |
13 Pedigree Analysis & Genetic Counseling
Pedigree analysis is the cornerstone of clinical genetics. Recognizing inheritance patterns from family histories allows proper diagnosis, risk calculation, and counseling.
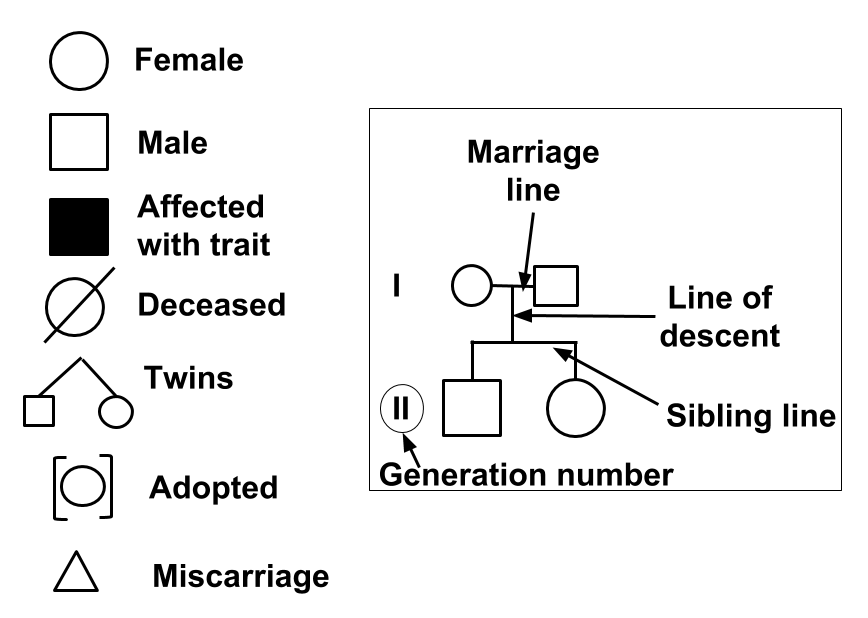
Pedigree Pattern Recognition
| Pattern | Key Clues | Risk to Offspring |
|---|---|---|
| Autosomal dominant | Vertical transmission; both sexes; male-to-male possible | 50% if one parent affected |
| Autosomal recessive | Horizontal; unaffected parents; consanguinity | 25% if both parents carriers |
| X-linked recessive | Affected males; carrier females; no male-to-male | 50% of sons affected (carrier mother) |
| X-linked dominant | Affected females > males; affected father → all daughters affected | 50% all children (affected mother) |
| Mitochondrial | Maternal transmission only; variable expressivity | All children of affected mother at risk |
Bayesian Analysis in Genetics
Bayesian analysis combines prior probability (based on pedigree) with conditional probability (based on additional information, such as test results or number of unaffected children) to calculate posterior probability. This is frequently tested in the context of calculating carrier risk for X-linked disorders.
Genetic counseling is non-directive — the counselor provides information and support but does not tell patients what decisions to make regarding testing, reproduction, or management. Key elements include: accurate diagnosis, inheritance pattern determination, recurrence risk calculation, discussion of testing options, psychosocial support, and informed consent.
Bayesian Analysis Example
A classic board scenario: A woman’s brother has hemophilia A (XLR). Her mother is an obligate carrier. She wants to know her risk of being a carrier, given that she has two unaffected sons.
| Carrier (prior = 1/2) | Non-carrier (prior = 1/2) | |
|---|---|---|
| Prior probability | 1/2 | 1/2 |
| Conditional probability (2 unaffected sons) | (1/2)² = 1/4 | 1 (certainty) |
| Joint probability | 1/2 × 1/4 = 1/8 | 1/2 × 1 = 1/2 |
| Posterior probability | (1/8) / (1/8 + 1/2) = 1/5 | (1/2) / (1/8 + 1/2) = 4/5 |
Her carrier risk decreases from 1/2 to 1/5 after having two unaffected sons. Each additional unaffected son further reduces her posterior carrier probability.
14 Trinucleotide Repeat Disorders & Anticipation
Trinucleotide repeat expansions are unstable mutations in which a three-nucleotide sequence is repeated an abnormal number of times. These repeats tend to expand during meiosis, leading to anticipation — earlier onset and/or more severe disease in successive generations.
Major Trinucleotide Repeat Disorders
| Disorder | Repeat | Location | Inheritance | Key Features |
|---|---|---|---|---|
| Huntington disease | CAG | Coding (huntingtin) | AD | Chorea, dementia, psychiatric symptoms; onset ~40; caudate atrophy; paternal anticipation |
| Fragile X syndrome | CGG | 5′ UTR (FMR1) | XLD | Most common inherited intellectual disability; long face, large ears, macroorchidism; maternal anticipation |
| Myotonic dystrophy type 1 | CTG | 3′ UTR (DMPK) | AD | Myotonia, distal muscle wasting, cataracts, cardiac arrhythmias, frontal balding; maternal anticipation (congenital form) |
| Friedreich ataxia | GAA | Intron (FXN/frataxin) | AR | Progressive ataxia, hypertrophic cardiomyopathy, diabetes; iron accumulation in mitochondria |
| Spinocerebellar ataxias | CAG | Coding | AD | Progressive cerebellar ataxia; multiple subtypes |
The FMR1 gene on Xq27.3 has CGG repeats. Normal: 5–44 repeats. Premutation: 55–200 repeats (carriers — males may develop fragile X-associated tremor/ataxia syndrome [FXTAS]; females may have premature ovarian insufficiency). Full mutation: >200 repeats → hypermethylation and silencing of FMR1 → absent FMRP protein. Expansion from premutation to full mutation occurs only during maternal transmission (oogenesis).
15 Genomic Imprinting & Uniparental Disomy
Genomic imprinting is the epigenetic silencing of one parental allele, so that only the allele from the other parent is expressed. This means loss or mutation of the active allele cannot be compensated by the silenced allele.
Imprinting Disorders
| Disorder | Chromosome | Mechanism | Key Features |
|---|---|---|---|
| Prader–Willi syndrome | 15q11–13 | Loss of paternal allele (maternal allele is imprinted/silenced) | Hypotonia at birth, hyperphagia → obesity, intellectual disability, hypogonadism, short stature |
| Angelman syndrome | 15q11–13 | Loss of maternal allele (UBE3A; paternal allele silenced in brain) | “Happy puppet” — severe intellectual disability, seizures, ataxic gait, inappropriate laughter |
| Beckwith–Wiedemann | 11p15.5 | Overexpression of paternal IGF2 or loss of maternal CDKN1C | Macrosomia, macroglossia, omphalocele, neonatal hypoglycemia, hemihypertrophy, Wilms tumor risk |
| Russell–Silver syndrome | 11p15.5 / chr 7 | Loss of paternal IGF2 or maternal UPD7 | Intrauterine and postnatal growth restriction, limb asymmetry, triangular face |
UPD occurs when both copies of a chromosome (or chromosomal region) come from one parent. Maternal UPD 15 causes Prader–Willi syndrome (two maternal copies, both silenced at the imprinted locus). Paternal UPD 15 causes Angelman syndrome. UPD can also unmask AR disorders if the parent contributing both copies is a carrier (isodisomy).
16 Mosaicism, Incomplete Penetrance & Variable Expressivity
Mosaicism
Mosaicism refers to the presence of two or more genetically distinct cell populations in a single individual, arising from a post-zygotic mutation.
- Somatic mosaicism — mutation in somatic cells; affects only tissues derived from the mutant clone; not transmitted to offspring
- Germline (gonadal) mosaicism — mutation in germ cells; phenotypically normal parent can transmit a mutation to multiple offspring; explains recurrence of “de novo” disorders in siblings (e.g., osteogenesis imperfecta)
- Confined placental mosaicism — abnormal karyotype in placenta but normal fetus; can cause false-positive CVS results
Incomplete Penetrance vs. Variable Expressivity
| Concept | Definition | Example |
|---|---|---|
| Incomplete penetrance | Not all individuals with a pathogenic genotype show the phenotype | BRCA1: ~70% lifetime risk of breast cancer (not 100%); some carriers never develop cancer |
| Variable expressivity | Individuals with the same genotype show different severity or features | NF1: one patient has only café-au-lait spots while a sibling has plexiform neurofibromas and optic glioma |
Other Non-Mendelian Concepts
- Locus heterogeneity — mutations in different genes produce the same phenotype (e.g., retinitis pigmentosa has >80 associated genes)
- Allelic heterogeneity — different mutations in the same gene produce the same disorder (e.g., >2,000 CFTR mutations all cause CF)
- Phenocopy — an environmentally caused phenotype that mimics a genetic disorder (e.g., thalidomide-induced phocomelia resembling Holt–Oram syndrome)
- Compound heterozygote — two different pathogenic alleles at the same locus (common in AR disorders like CF)
Multifactorial Inheritance
Multifactorial (complex) disorders result from the combined effects of multiple genes and environmental factors. They do not follow Mendelian patterns but show familial clustering.
| Feature | Multifactorial | Mendelian |
|---|---|---|
| Inheritance pattern | No clear pattern; familial clustering | Predictable ratios (AD, AR, XL) |
| Recurrence risk | Empiric (2–5% for first-degree relatives) | Calculated from genotype (25%, 50%) |
| Concordance in MZ twins | <100% (typically 20–60%) | ~100% for fully penetrant conditions |
| Environmental influence | Significant | Minimal (for highly penetrant alleles) |
Threshold Model
The liability threshold model explains multifactorial disorders with a discontinuous phenotype (present or absent). Individuals exceeding a threshold of combined genetic and environmental liability develop the condition.
- Risk increases with the number of affected relatives
- Risk increases with severity of the condition in the proband
- Risk is higher when the affected individual is of the less commonly affected sex (e.g., pyloric stenosis is more common in males; a female proband suggests higher genetic load, so her relatives have higher recurrence risk)
Common Multifactorial Conditions
- Neural tube defects — anencephaly, spina bifida; risk reduced by periconceptional folic acid supplementation
- Congenital heart defects — most common birth defects; VSD most frequent
- Cleft lip/palate — recurrence risk ~4% for first-degree relatives
- Type 2 diabetes mellitus — strong genetic component; HLA associations less prominent than in type 1
- Coronary artery disease — polygenic risk scores increasingly used clinically
- Pyloric stenosis — 5:1 male predominance; threshold model example
17 Chromosomal Structure & Aberrations
Chromosomal aberrations are changes in chromosome number or structure visible by cytogenetic techniques. They are a major cause of congenital anomalies, intellectual disability, and pregnancy loss.
Structural Aberrations
| Type | Description | Clinical Significance |
|---|---|---|
| Deletion | Loss of a chromosomal segment | Cri-du-chat (5p−); Williams syndrome (7q11.23); DiGeorge/velocardiofacial (22q11.2) |
| Duplication | Extra copy of a chromosomal segment | Generally less severe than deletions; Charcot–Marie–Tooth 1A (17p duplication) |
| Inversion | Segment reversed in orientation (paracentric or pericentric) | Usually balanced (no phenotypic effect) but offspring at risk for unbalanced gametes |
| Translocation (reciprocal) | Exchange of segments between non-homologous chromosomes | Balanced carriers phenotypically normal; offspring at risk for unbalanced karyotype |
| Robertsonian translocation | Fusion of two acrocentric chromosomes (13, 14, 15, 21, 22) at centromeres | Most common: rob(14;21) → risk of translocation Down syndrome; 45 chromosomes in carrier |
| Isochromosome | Chromosome with two identical arms (duplication of one arm + deletion of other) | Isochromosome Xq is seen in some Turner syndrome patients |
| Ring chromosome | Circular chromosome from deletion of both telomeric ends and fusion | Variable phenotype depending on deleted material |
Microdeletion Syndromes
| Syndrome | Deletion | Key Features |
|---|---|---|
| DiGeorge / 22q11.2 deletion | 22q11.2 | CATCH-22: Cardiac defects, Abnormal facies, Thymic aplasia, Cleft palate, Hypocalcemia (absent parathyroids) |
| Williams syndrome | 7q11.23 (elastin gene) | “Elfin” facies, supravalvular aortic stenosis, intellectual disability with overly friendly personality, hypercalcemia |
| Cri-du-chat | 5p− | High-pitched cat-like cry, microcephaly, intellectual disability |
| Wolf–Hirschhorn | 4p− | “Greek warrior helmet” facies, growth restriction, seizures, intellectual disability |
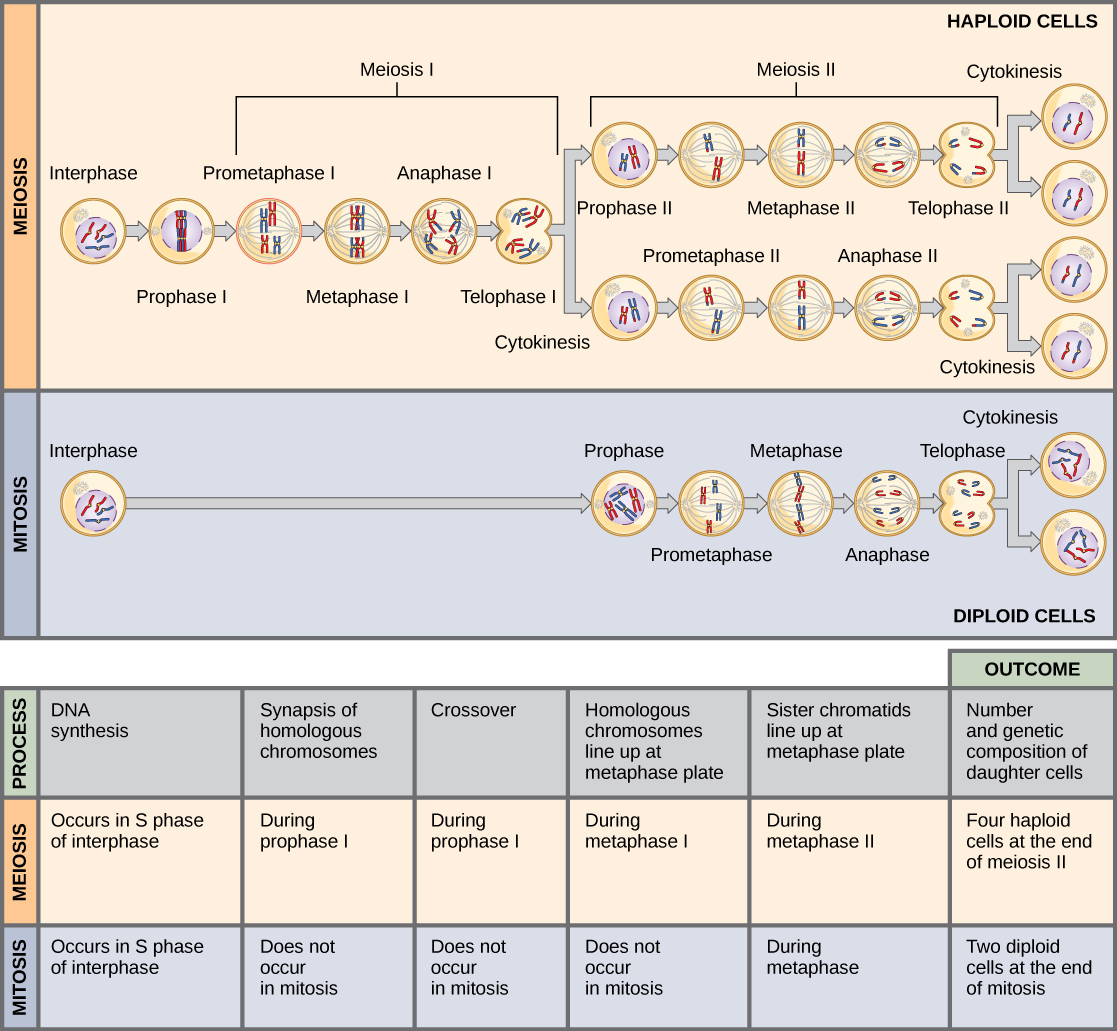
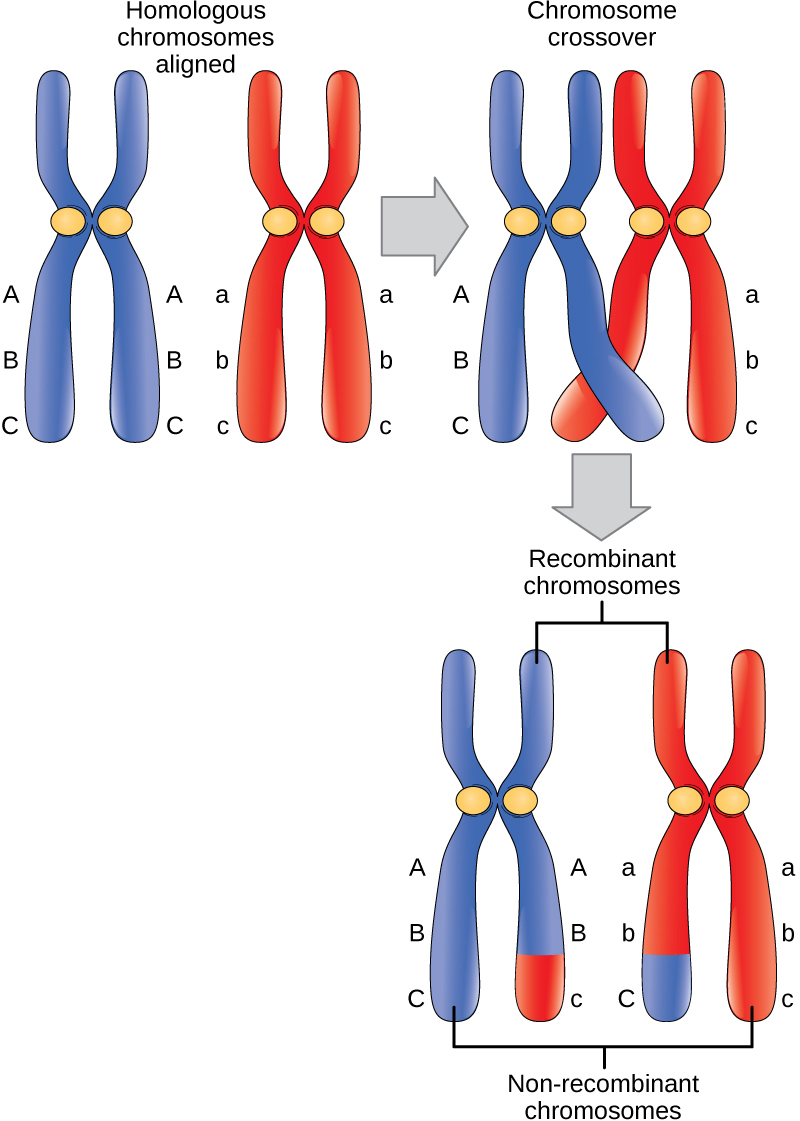
Meiosis and Nondisjunction
Understanding the stages of meiosis is critical for explaining how aneuploidies arise.
| Stage | Events | Consequence of Nondisjunction |
|---|---|---|
| Meiosis I | Homologous chromosomes separate; crossing over occurs in prophase I | Both homologs go to one daughter cell → gametes with n+1 or n−1; most common cause of trisomy 21 |
| Meiosis II | Sister chromatids separate (similar to mitosis) | Both chromatids go to one cell; two normal gametes + one n+1 + one n−1 |
| Mitotic nondisjunction | Post-zygotic error in cell division | Produces mosaicism (some cells normal, some aneuploid) |
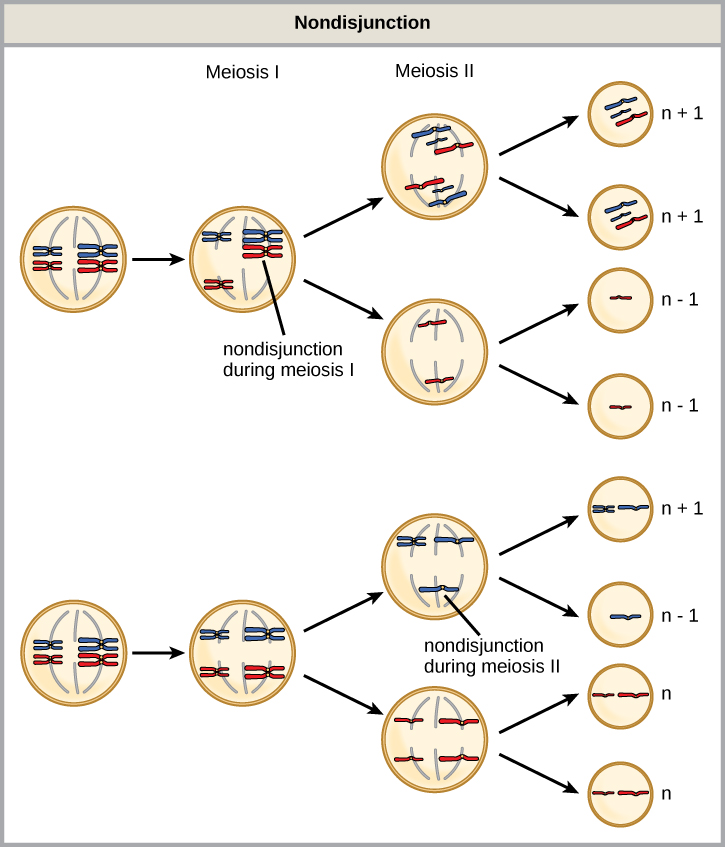
18 Autosomal Aneuploidies
Aneuploidy is an abnormal chromosome number that is not a multiple of the haploid set (n = 23). It most commonly results from nondisjunction during meiosis I or meiosis II. Advanced maternal age is the primary risk factor.
Major Autosomal Aneuploidies
| Disorder | Karyotype | Key Features | Associations |
|---|---|---|---|
| Down syndrome (trisomy 21) | 47,XX/XY,+21 | Flat facies, epicanthal folds, single palmar crease, intellectual disability, hypotonia | ASD/VSD, duodenal atresia, Hirschsprung disease, ALL, early-onset Alzheimer (~40s), atlantoaxial instability |
| Edwards syndrome (trisomy 18) | 47,XX/XY,+18 | Rocker-bottom feet, clenched fists (overlapping fingers), micrognathia, prominent occiput | VSD, horseshoe kidney; death usually by age 1; 2nd most common autosomal trisomy |
| Patau syndrome (trisomy 13) | 47,XX/XY,+13 | Holoprosencephaly, cleft lip/palate, polydactyly, microphthalmia, cutis aplasia | Severe intellectual disability; death usually by age 1 |
95% of cases result from meiotic nondisjunction (maternal meiosis I in most cases; risk increases with maternal age). ~4% from Robertsonian translocation, most commonly rob(14;21) — the translocation carrier parent has 45 chromosomes but is phenotypically normal; recurrence risk depends on which parent carries the translocation. ~1% from mosaicism (milder phenotype).
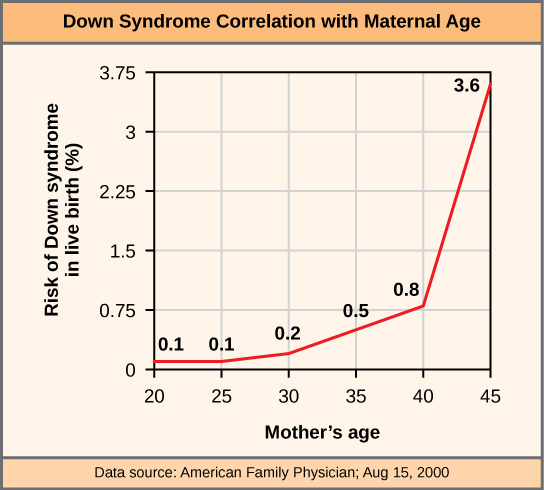
Prenatal Screening for Aneuploidies
| Trimester | Test | Markers |
|---|---|---|
| First | Combined screening | ↓ PAPP-A, ↑ free β-hCG, ↑ nuchal translucency (Down); opposite pattern for trisomy 18 |
| Second | Quad screen | ↓ AFP, ↑ β-hCG, ↓ estriol, ↑ inhibin A (Down); all low in trisomy 18; ↑ AFP in neural tube defects |
| Any gestational age (≥10 wk) | Cell-free fetal DNA (NIPT) | Analyzes placental DNA in maternal blood; high sensitivity/specificity for trisomies 21, 18, 13 |
19 Sex Chromosome Disorders
Turner Syndrome (45,X)
The only monosomy compatible with life. Affects females. Most 45,X conceptions end in spontaneous abortion (99%); live-born incidence is ~1/2,500 female births.
- Short stature, shield chest, widely spaced nipples, webbed neck (cystic hygroma remnant)
- Primary amenorrhea, streak gonads, infertility
- Coarctation of the aorta (preductal), bicuspid aortic valve
- Horseshoe kidney, lymphedema at birth
- Normal intelligence (may have difficulties with spatial processing)
- No Barr bodies (only one X chromosome)
Klinefelter Syndrome (47,XXY)
Affects males. Incidence ~1/600 male births. Most common cause of hypogonadism and infertility in males.
- Tall stature, long extremities (eunuchoid body habitus)
- Testicular atrophy, infertility (dysgenesis of seminiferous tubules)
- Gynecomastia, decreased facial/body hair
- ↑ FSH, ↑ LH, ↑ estrogen, ↓ testosterone
- One Barr body (inactivated extra X)
- Mild intellectual disability (verbal); increased risk of breast cancer and SLE
Other Sex Chromosome Conditions
| Karyotype | Phenotype | Key Features |
|---|---|---|
| 47,XYY | Male | Tall stature, severe acne, normal fertility; historically (incorrectly) associated with aggression; normal phenotype in most |
| 47,XXX (triple X) | Female | Tall stature, usually clinically normal; may have mild learning difficulties; most undiagnosed |
| 46,XX testicular DSD | Phenotypic male | SRY gene translocated to X chromosome; male phenotype with small testes |
| 45,X/46,XY mosaicism | Variable | Mixed gonadal dysgenesis; phenotype ranges from Turner-like to ambiguous genitalia to normal male |
In females, one X chromosome is randomly inactivated early in embryogenesis to achieve dosage compensation. The inactive X forms the Barr body. Number of Barr bodies = number of X chromosomes minus 1. XIST RNA (from the inactive X) coats the chromosome and recruits silencing complexes. Skewed X-inactivation can cause carrier females to manifest X-linked recessive conditions.
Disorders of Sexual Development (DSD)
| Condition | Karyotype | Mechanism | Phenotype |
|---|---|---|---|
| Complete androgen insensitivity | 46,XY | Nonfunctional androgen receptor (XLR) | Female external genitalia, absent uterus, blind vaginal pouch, undescended testes, breast development at puberty (estrogen from aromatization); raised female |
| 5α-reductase deficiency | 46,XY | Cannot convert testosterone to DHT (AR) | Ambiguous genitalia at birth; virilization at puberty (testosterone surge); “guevedoces” |
| Congenital adrenal hyperplasia | 46,XX | 21-hydroxylase deficiency (most common); excess androgens | Virilized female (ambiguous genitalia); salt-wasting in severe form; elevated 17-OH progesterone |
| Swyer syndrome | 46,XY | SRY mutation → no testis development | Female phenotype with streak gonads; no puberty; gonadoblastoma risk |
| True gonadal DSD | Variable | Both ovarian and testicular tissue present | Ambiguous genitalia; ovotestis |
20 Oncogenes & Tumor Suppressors
Cancer is fundamentally a genetic disease arising from accumulated mutations in genes controlling cell growth, differentiation, and death. Two major categories of cancer genes exist: oncogenes (gain-of-function, dominant) and tumor suppressors (loss-of-function, recessive at the cellular level).
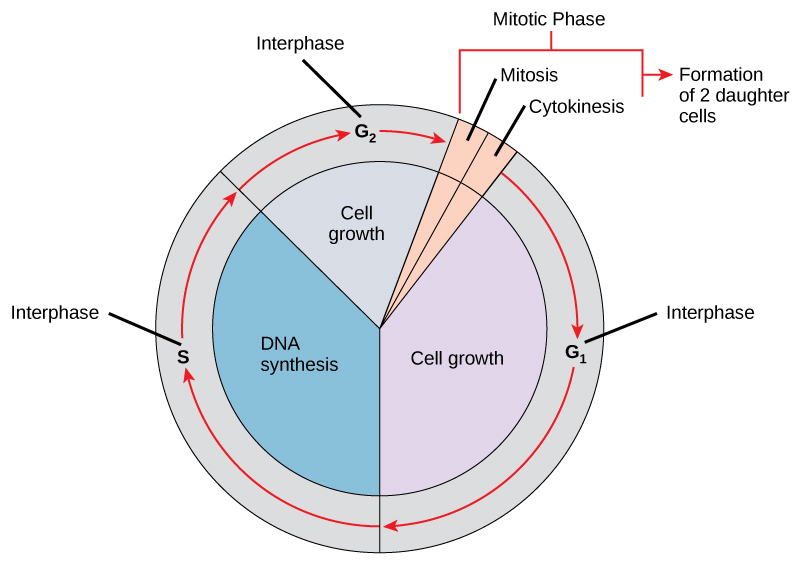
Cell Cycle Review
| Phase | Events | Key Regulators |
|---|---|---|
| G1 | Cell growth; preparation for DNA synthesis | Cyclin D + CDK4/6; restriction point; Rb hypophosphorylation holds cell in G1 |
| S | DNA synthesis (replication) | Cyclin E + CDK2 (G1/S transition); Cyclin A + CDK2 |
| G2 | Preparation for mitosis; error checking | Cyclin A + CDK1; DNA damage checkpoint (ATM/ATR, Chk1/2) |
| M | Mitosis (prophase, metaphase, anaphase, telophase) + cytokinesis | Cyclin B + CDK1 (MPF); spindle assembly checkpoint |
CDK Inhibitors (Tumor Suppressors)
- p21 (CDKN1A) — induced by p53; universal CDK inhibitor; mediates G1 arrest after DNA damage
- p27 (CDKN1B) — inhibits cyclin E-CDK2; regulates G1/S; loss associated with poor cancer prognosis
- p16 (CDKN2A/INK4a) — specifically inhibits CDK4/6; frequently deleted or silenced in cancers; same locus encodes ARF (p14) which stabilizes p53
Proto-Oncogenes → Oncogenes
Proto-oncogenes are normal genes that promote cell growth. A single activating mutation converts them to oncogenes (gain-of-function, dominant). Mechanisms include point mutation, gene amplification, and chromosomal translocation.
| Oncogene | Normal Function | Activation | Associated Cancer |
|---|---|---|---|
| RAS (KRAS, HRAS, NRAS) | GTPase signal transduction | Point mutation (constitutively active) | Pancreatic, colon, lung adenocarcinoma |
| HER2/neu (ERBB2) | Receptor tyrosine kinase | Gene amplification | Breast cancer (~20%); targeted by trastuzumab |
| c-MYC | Transcription factor | Translocation t(8;14) | Burkitt lymphoma |
| BCL-2 | Anti-apoptotic protein | Translocation t(14;18) | Follicular lymphoma |
| ABL | Tyrosine kinase | Translocation t(9;22) → BCR-ABL | CML (Philadelphia chromosome); targeted by imatinib |
| RET | Receptor tyrosine kinase | Point mutation | MEN 2A, 2B; medullary thyroid carcinoma |
| BRAF | Serine/threonine kinase (MAPK pathway) | Point mutation (V600E) | Melanoma, hairy cell leukemia, papillary thyroid cancer |
| KIT | Receptor tyrosine kinase | Activating mutation | Gastrointestinal stromal tumor (GIST); targeted by imatinib |
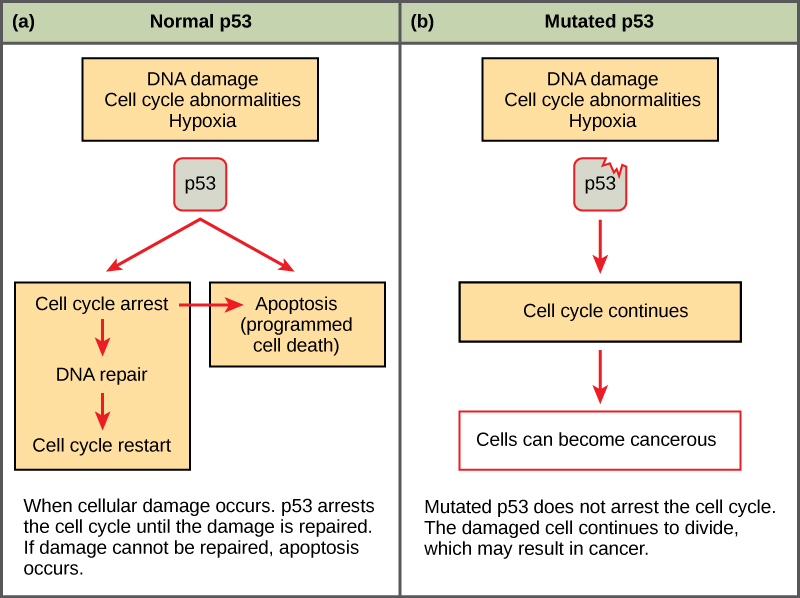
Tumor Suppressor Genes
Tumor suppressors inhibit cell proliferation or promote apoptosis. Both alleles must be inactivated for loss of function (Knudson’s two-hit hypothesis). In hereditary cancer syndromes, one hit is inherited (germline) and the second is somatic.
| Gene | Function | Associated Syndrome / Cancer |
|---|---|---|
| TP53 | “Guardian of the genome” — activates p21 (CDK inhibitor), promotes DNA repair, induces apoptosis | Li–Fraumeni syndrome; most commonly mutated gene in human cancers |
| RB1 | Cell cycle checkpoint (G1→S); phosphorylated by CDK4/6 | Retinoblastoma; osteosarcoma |
| BRCA1 / BRCA2 | Homologous recombination DNA repair | Hereditary breast and ovarian cancer; BRCA2 also prostate and pancreatic |
| APC | Wnt signaling inhibitor (β-catenin degradation) | Familial adenomatous polyposis (FAP); colorectal cancer |
| VHL | Targets HIF for degradation under normoxic conditions | Von Hippel–Lindau syndrome; clear cell renal carcinoma, hemangioblastomas, pheochromocytoma |
| WT1 | Transcription factor (kidney development) | Wilms tumor (WAGR syndrome, Denys–Drash) |
| NF1 | Neurofibromin (RAS-GAP; inactivates RAS) | Neurofibromatosis type 1 |
| NF2 | Merlin (cytoskeletal protein) | Neurofibromatosis type 2; bilateral acoustic schwannomas |
| PTEN | Phosphatase (antagonizes PI3K/AKT pathway) | Cowden syndrome; hamartomas, breast/thyroid/endometrial cancer |
High-Yield Chromosomal Translocations in Cancer
| Translocation | Genes Involved | Cancer | Notes |
|---|---|---|---|
| t(9;22) | BCR-ABL | CML; also Ph+ ALL | Philadelphia chromosome; constitutive tyrosine kinase; imatinib |
| t(8;14) | c-MYC / IgH | Burkitt lymphoma | “Starry sky” pattern; EBV association; jaw mass in endemic form |
| t(14;18) | BCL-2 / IgH | Follicular lymphoma | Overexpression of anti-apoptotic BCL-2; indolent course |
| t(11;14) | Cyclin D1 / IgH | Mantle cell lymphoma | Overexpression of cyclin D1; aggressive |
| t(15;17) | PML-RARα | Acute promyelocytic leukemia (M3) | Responds to ATRA (all-trans retinoic acid) + arsenic trioxide; Auer rods, DIC |
| t(11;22) | EWS-FLI1 | Ewing sarcoma | Small round blue cell tumor of bone in children/adolescents |
| t(12;21) | ETV6-RUNX1 | Childhood ALL (B-cell) | Most common translocation in pediatric ALL; good prognosis |
21 Hereditary Cancer Syndromes
Hereditary cancer syndromes account for 5–10% of all cancers. They result from germline mutations in tumor suppressor genes or DNA repair genes and follow an autosomal dominant inheritance pattern with incomplete penetrance.
Major Hereditary Cancer Syndromes
| Syndrome | Gene | Cancers / Features |
|---|---|---|
| Lynch syndrome (HNPCC) | MLH1, MSH2, MSH6, PMS2 | Colorectal (right-sided), endometrial, ovarian, gastric, urinary tract; microsatellite instability |
| Familial adenomatous polyposis | APC | Hundreds to thousands of colonic polyps by adolescence; 100% CRC risk if untreated; Gardner variant (osteomas, desmoids) |
| Li–Fraumeni syndrome | TP53 | Sarcomas, breast cancer, leukemia, brain tumors, adrenocortical carcinoma (SBLA cancers); multiple primary cancers |
| Hereditary breast/ovarian cancer | BRCA1, BRCA2 | Breast (bilateral, early onset), ovarian, prostate, pancreatic; PARP inhibitors effective |
| Von Hippel–Lindau | VHL | Hemangioblastomas (cerebellum, retina), clear cell RCC, pheochromocytoma |
| MEN 1 | MEN1 (menin) | Parathyroid, pituitary, pancreatic tumors (“3 P’s”) |
| MEN 2A | RET | Medullary thyroid carcinoma, pheochromocytoma, parathyroid hyperplasia |
| MEN 2B | RET | Medullary thyroid carcinoma, pheochromocytoma, mucosal neuromas, marfanoid habitus |
| Cowden syndrome | PTEN | Hamartomas, increased risk of breast, thyroid, and endometrial cancer; trichilemmomas |
| Peutz–Jeghers | STK11/LKB1 | Hamartomatous GI polyps, mucocutaneous hyperpigmentation, increased GI and non-GI cancer risk |
Amsterdam II criteria require ≥3 relatives with Lynch-associated cancers (one a first-degree relative of the other two), ≥2 successive generations, and ≥1 diagnosed before age 50. Universal screening of all CRC with immunohistochemistry for MMR proteins (MLH1, MSH2, MSH6, PMS2) or microsatellite instability (MSI) testing is now recommended to identify Lynch syndrome patients.
22 Population Genetics & Hardy–Weinberg Equilibrium
Population genetics studies allele and genotype frequencies in populations and the forces that change them over time.
Hardy–Weinberg Equilibrium
In a large, randomly mating population with no selection, mutation, migration, or genetic drift, allele and genotype frequencies remain constant across generations.
For a locus with two alleles (p = frequency of dominant allele, q = frequency of recessive allele):
- p + q = 1
- p² + 2pq + q² = 1
- p² = frequency of homozygous dominant (AA)
- 2pq = frequency of heterozygous carriers (Aa)
- q² = frequency of homozygous recessive (affected, aa)
Applying Hardy–Weinberg
Board-style application: If an AR disease has a prevalence of 1/10,000, then:
- q² = 1/10,000 → q = 1/100
- p = 1 − 1/100 = 99/100 ≈ 1
- Carrier frequency (2pq) = 2 × 1 × 1/100 = 1/50
Forces That Disrupt Hardy–Weinberg Equilibrium
| Force | Effect | Example |
|---|---|---|
| Natural selection | Differential survival/reproduction based on genotype | Sickle cell heterozygote advantage in malaria-endemic regions |
| Genetic drift | Random fluctuation in allele frequencies in small populations | Founder effect; bottleneck effect |
| Founder effect | High frequency of an allele in a population descended from a small founding group | Tay–Sachs in Ashkenazi Jews; Ellis–van Creveld in Amish |
| Non-random mating | Assortative mating or consanguinity | Consanguinity increases homozygosity → more AR disorders |
| Migration (gene flow) | Introduction of new alleles into a population | Changes allele frequencies toward migrant source |
| Mutation | Introduces new alleles | New mutations in tumor suppressor genes |
Heterozygote Advantage
Balanced polymorphism: heterozygous carriers of certain AR conditions have a selective advantage, maintaining the disease allele at a higher frequency than expected.
| Disease | Heterozygote Advantage Against |
|---|---|
| Sickle cell trait (HbAS) | Plasmodium falciparum malaria |
| Thalassemia trait | Malaria |
| G6PD deficiency | Malaria |
| Cystic fibrosis carrier | Cholera / typhoid fever (proposed) |
| CCR5-Δ32 heterozygote | HIV infection |
Genetic Epidemiology Measures
| Measure | Definition | How Estimated |
|---|---|---|
| Heritability (h²) | Proportion of phenotypic variation attributable to genetic factors | Twin studies: h² = 2(rMZ − rDZ) where r = concordance rate |
| Concordance rate | Probability that both twins are affected when one is affected | Higher MZ than DZ concordance suggests genetic contribution |
| Relative risk (λ) | Risk in relatives of affected individual / population risk | λs (sibling recurrence risk ratio) used for complex traits |
Consanguinity and Inbreeding
The coefficient of inbreeding (F) is the probability that an individual is homozygous at a locus due to shared ancestry of the parents.
- First cousins: F = 1/16
- Second cousins: F = 1/64
- Parent-offspring or sibling: F = 1/4
Consanguinity increases the proportion of homozygous loci, thereby increasing the risk of autosomal recessive conditions. In populations with high consanguinity rates, AR disorders are more prevalent than predicted by Hardy–Weinberg (which assumes random mating).
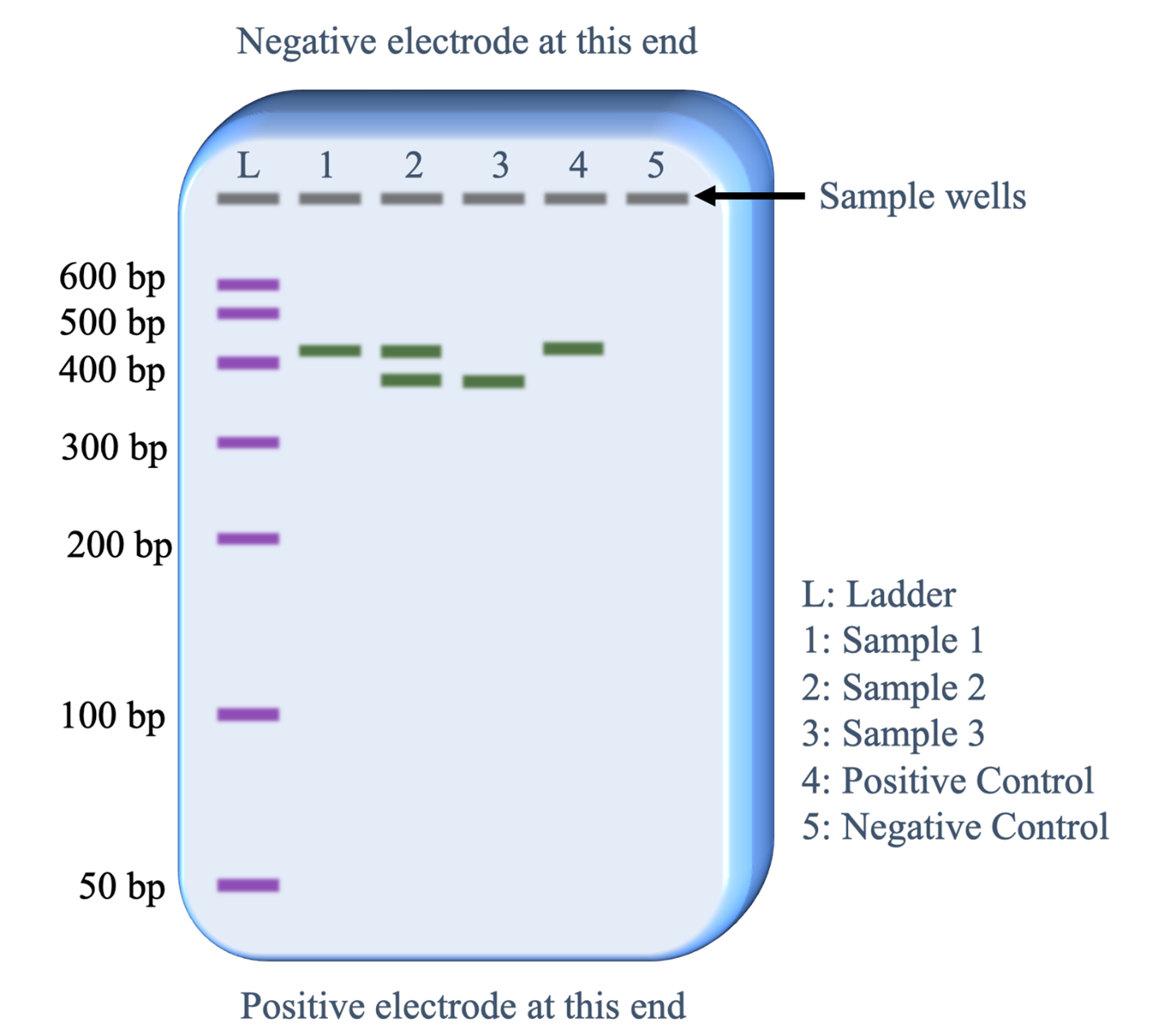
23 Genetic Testing & Molecular Diagnostics
Advances in molecular technology have transformed genetic diagnosis. Understanding the capabilities and limitations of each technique is essential for clinical practice and is frequently tested.
Major Genetic Testing Methods
| Technique | What It Detects | Clinical Applications |
|---|---|---|
| Karyotype | Chromosome number and gross structural abnormalities (≥5–10 Mb) | Aneuploidies (trisomy 21), translocations, large deletions; requires dividing cells (PHA-stimulated lymphocytes) |
| FISH | Specific known deletions, duplications, translocations | Microdeletion syndromes (22q11.2), BCR-ABL, HER2 amplification; rapid (no need for dividing cells) |
| Chromosomal microarray (CMA/CGH) | Genome-wide copy number variations (CNVs) at high resolution | First-line test for intellectual disability and multiple congenital anomalies; detects submicroscopic deletions/duplications; cannot detect balanced translocations |
| PCR | Amplification of specific DNA sequences | Detect known point mutations, trinucleotide repeat sizing, pathogen detection, STR analysis (paternity) |
| Sanger sequencing | Single gene sequencing (gold standard for single variants) | Confirm specific mutations; limited throughput |
| Next-generation sequencing (NGS) | Massively parallel sequencing of gene panels, whole exome, or whole genome | Multigene panels for hereditary cancer; whole exome sequencing for undiagnosed disorders; tumor profiling |
| Southern blot | Specific DNA sequences; size of DNA fragments | Trinucleotide repeat expansions (fragile X); largely replaced by PCR/NGS |
| Western blot | Specific proteins | Dystrophin in muscular dystrophy; confirmatory HIV testing (historical) |
| Northern blot | Specific RNA species | Research tool; largely replaced by RT-PCR |
SNoW DRoP: Southern = DNA; Northern = RNA; Western = Protein. Southern blot was named after its inventor (Edwin Southern); Northern and Western were named by analogy.
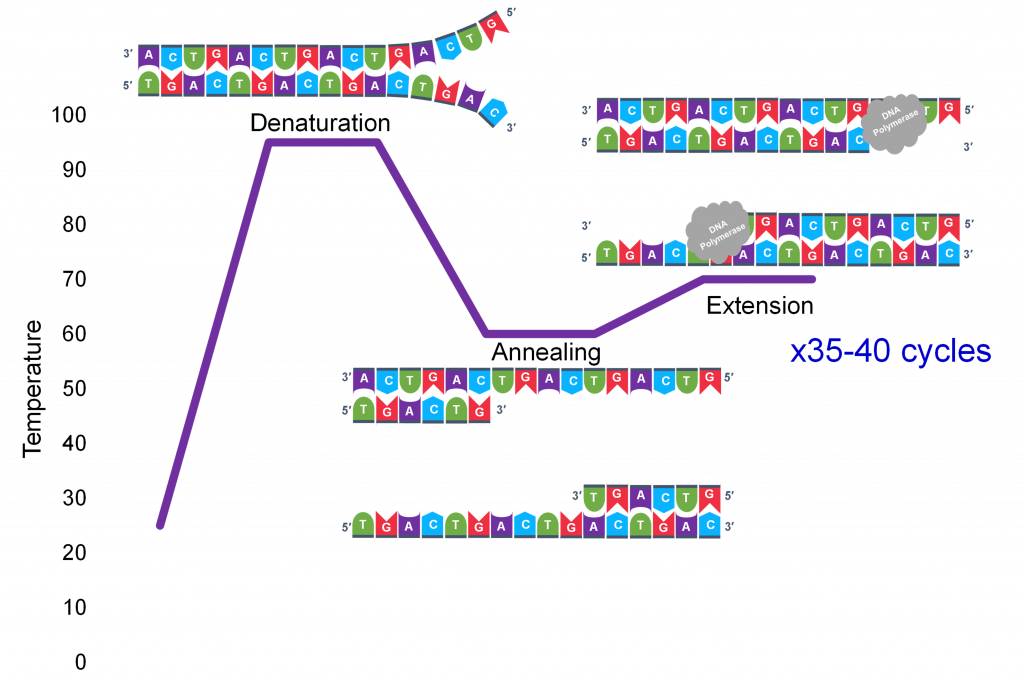
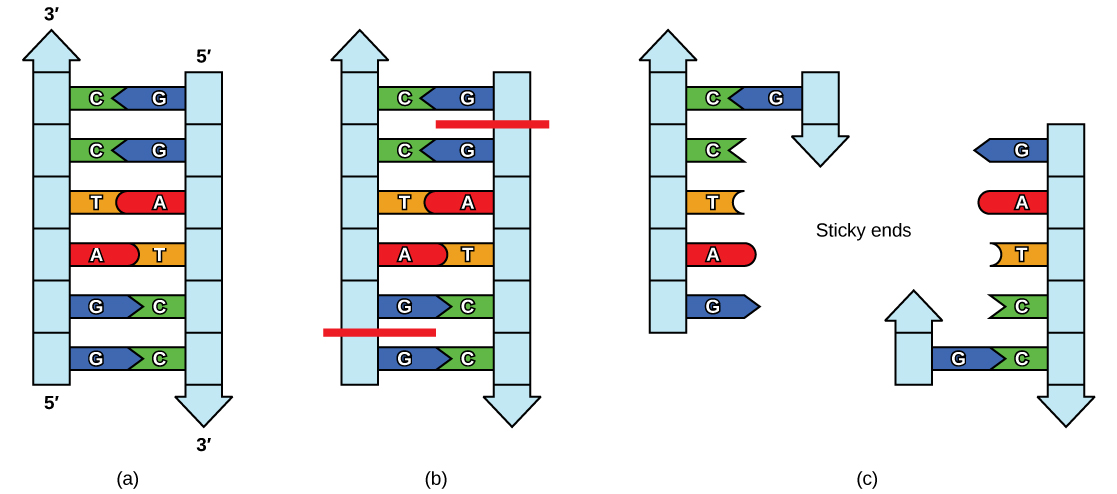
Gel Electrophoresis & Restriction Enzymes
- Restriction endonucleases — cut DNA at specific palindromic sequences; create restriction fragment length polymorphisms (RFLPs) useful for genetic mapping
- Gel electrophoresis — separates DNA/RNA/protein fragments by size (smaller fragments migrate faster toward the anode); DNA is negatively charged (phosphate backbone)
PCR Variants
| Technique | Application |
|---|---|
| Standard PCR | Amplify specific DNA sequences for analysis |
| RT-PCR | Reverse transcriptase converts mRNA to cDNA, then amplify; detects gene expression |
| qPCR (real-time) | Quantitative measurement of DNA/RNA; viral load monitoring (HIV, HCV) |
| Multiplex PCR | Multiple primer sets in one reaction; used in DMD deletion screening |
| Allele-specific PCR | Distinguishes between specific alleles; used for known point mutations (e.g., Factor V Leiden) |
| Methylation-specific PCR | Detects DNA methylation status; used for imprinting disorders (Prader–Willi/Angelman) |
Clinical Genetic Testing Approach
- Known familial mutation → targeted PCR/Sanger sequencing
- Suspected chromosomal abnormality → karyotype (gross) or CMA (submicroscopic)
- Suspected microdeletion → FISH or CMA
- Undiagnosed genetic condition → whole exome/genome sequencing
- Prenatal aneuploidy screening → cell-free fetal DNA (NIPT), followed by amniocentesis/CVS for confirmation
24 Pharmacogenomics
Pharmacogenomics studies how genetic variation affects drug metabolism, efficacy, and toxicity. Individualizing drug therapy based on genotype is a cornerstone of precision medicine.
Cytochrome P450 Polymorphisms
| Enzyme | Clinical Significance | Substrate Examples |
|---|---|---|
| CYP2D6 | Poor metabolizers: toxicity from codeine (no conversion to morphine), tamoxifen (reduced efficacy); ultra-rapid metabolizers: excess morphine from codeine → respiratory depression | Codeine, tamoxifen, fluoxetine, metoprolol |
| CYP2C19 | Poor metabolizers: increased clopidogrel failure (prodrug, not activated); omeprazole accumulation | Clopidogrel, omeprazole, voriconazole |
| CYP2C9 | Poor metabolizers: increased warfarin sensitivity; dose reduction needed | Warfarin, phenytoin, NSAIDs |
| CYP3A4 | Metabolizes ~50% of all drugs; inhibited by grapefruit juice, azole antifungals, macrolides | Statins, cyclosporine, tacrolimus, many others |
Other Pharmacogenomic Associations
| Genetic Variant | Drug | Clinical Implication |
|---|---|---|
| HLA-B*5701 | Abacavir | Severe hypersensitivity reaction; test before prescribing |
| HLA-B*1502 | Carbamazepine | Stevens–Johnson syndrome / toxic epidermal necrolysis in Southeast Asian populations |
| HLA-B*5801 | Allopurinol | Severe cutaneous adverse reactions; common in Southeast Asian and African American populations |
| VKORC1 variants | Warfarin | Affect vitamin K epoxide reductase sensitivity; determine warfarin dose requirement |
| TPMT deficiency | 6-mercaptopurine, azathioprine | Thiopurine methyltransferase deficiency → severe myelosuppression; dose reduce or avoid |
| UGT1A1*28 (Gilbert) | Irinotecan | Reduced glucuronidation of SN-38 (active metabolite) → severe diarrhea, neutropenia |
| G6PD deficiency | Primaquine, dapsone, sulfonamides | Oxidant stress → hemolytic anemia; test before prescribing antimalarials |
| NAT2 (slow acetylator) | Isoniazid, hydralazine, procainamide | Increased risk of drug-induced lupus (hydralazine, procainamide) and peripheral neuropathy (isoniazid) |
Phase I vs. Phase II Drug Metabolism
| Phase | Reactions | Key Enzymes | Genetic Variation Impact |
|---|---|---|---|
| Phase I | Oxidation, reduction, hydrolysis (add/expose functional groups) | Cytochrome P450 family (CYP2D6, CYP3A4, CYP2C9, CYP2C19) | Poor, intermediate, extensive, ultra-rapid metabolizer phenotypes |
| Phase II | Conjugation (glucuronidation, sulfation, acetylation, methylation, glutathione) | UGT1A1, NAT2, TPMT, COMT, GSTs | NAT2 slow acetylators: isoniazid toxicity; UGT1A1*28: irinotecan toxicity |
25 Gene Therapy & Emerging Technologies
Gene Therapy Approaches
| Approach | Mechanism | Examples |
|---|---|---|
| Gene replacement | Deliver functional copy of defective gene (via viral vector) | Luxturna (voretigene) for RPE65-associated retinal dystrophy; Zolgensma (onasemnogene) for SMA |
| Gene editing (CRISPR-Cas9) | Precise modification of endogenous DNA using guide RNA + Cas9 nuclease | Casgevy (exagamglogene) for sickle cell disease and β-thalassemia (first approved CRISPR therapy) |
| Antisense oligonucleotides | Bind mRNA to modulate splicing or block translation | Nusinersen (Spinraza) for SMA; eteplirsen for Duchenne (exon skipping) |
| RNA interference (siRNA) | Degrade target mRNA via RISC complex | Patisiran for hereditary transthyretin amyloidosis; inclisiran for hypercholesterolemia |
| CAR-T cell therapy | Patient T cells engineered to express chimeric antigen receptor | Tisagenlecleucel for B-ALL; axicabtagene for DLBCL |
Viral Vectors for Gene Delivery
| Vector | Advantages | Limitations |
|---|---|---|
| Adeno-associated virus (AAV) | Non-integrating; low immunogenicity; long-term expression in non-dividing cells | Limited cargo size (~4.7 kb); pre-existing immunity in some patients |
| Lentivirus | Integrates into host genome; can transduce non-dividing cells; larger cargo | Risk of insertional mutagenesis (though much lower than retrovirus) |
| Retrovirus | Stable integration; efficient transduction | Only transduces dividing cells; insertional mutagenesis risk (caused leukemia in early SCID-X1 trials) |
| Adenovirus | Large cargo capacity; high transduction efficiency | Strong immune response; transient expression; not commonly used for gene therapy now |
The CRISPR system uses a guide RNA (gRNA) complementary to the target DNA sequence to direct the Cas9 endonuclease to create a double-strand break at a precise location. The cell then repairs the break via NHEJ (gene disruption) or homology-directed repair (gene correction with a template). Off-target effects remain a concern in clinical applications.
26 Clinical Correlates Across Specialties
Genetics intersects with virtually every clinical specialty. Understanding these connections is essential for integrated clinical reasoning.
Specialty-Specific Genetic Considerations
| Specialty | Key Genetic Topics | Examples |
|---|---|---|
| Cardiology | Channelopathies, cardiomyopathies, familial hyperlipidemia | Long QT syndrome (KCNQ1, KCNH2, SCN5A); hypertrophic cardiomyopathy (MYH7, MYBPC3); Marfan aortic dissection |
| Oncology | Somatic tumor profiling, hereditary cancer syndromes | EGFR mutations in NSCLC (gefitinib); BRCA in ovarian cancer (olaparib); MSI-H tumors (pembrolizumab) |
| Neurology | Trinucleotide repeats, neuromuscular disorders | Huntington, Friedreich ataxia, Duchenne/Becker, SMA (SMN1 deletion) |
| Hematology | Hemoglobinopathies, coagulopathies, bone marrow failure | Sickle cell, thalassemias, hemophilia, Factor V Leiden, Fanconi anemia |
| Pediatrics | Inborn errors of metabolism, chromosomal disorders, newborn screening | PKU, galactosemia, Down syndrome, metabolic storage diseases |
| Obstetrics | Prenatal genetic testing, carrier screening, teratogenesis | NIPT, carrier screening for CF/SCD, CVS vs. amniocentesis |
Newborn Screening
All US states mandate newborn screening (NBS) for a core panel of conditions. Screening typically uses dried blood spot collected 24–48 hours after birth.
- Phenylketonuria (PKU) — elevated phenylalanine; dietary restriction prevents intellectual disability
- Congenital hypothyroidism — elevated TSH; early thyroxine replacement prevents cretinism
- Galactosemia — elevated galactose-1-phosphate; dietary galactose restriction
- Sickle cell disease — hemoglobin electrophoresis; penicillin prophylaxis
- Cystic fibrosis — elevated immunoreactive trypsinogen (IRT); confirmatory sweat chloride test
- Maple syrup urine disease — elevated branched-chain amino acids
- Biotinidase deficiency — enzyme assay; biotin supplementation
- CAH (21-hydroxylase deficiency) — elevated 17-hydroxyprogesterone
ACOG recommends offering carrier screening for cystic fibrosis and spinal muscular atrophy to all pregnant women (or those planning pregnancy), regardless of ethnicity. Expanded carrier screening panels that test for 100+ AR conditions simultaneously using NGS are increasingly used. Ashkenazi Jewish heritage warrants additional screening for Tay–Sachs, Gaucher, Canavan, familial dysautonomia, and others.
Prenatal Diagnostic Procedures
| Procedure | Timing | What It Provides | Risks / Notes |
|---|---|---|---|
| Chorionic villus sampling (CVS) | 10–13 weeks | Fetal karyotype, DNA analysis, enzyme assays | ~1% miscarriage risk; cannot detect neural tube defects; risk of confined placental mosaicism |
| Amniocentesis | 15–20 weeks | Fetal karyotype, AFP (neural tube defects), DNA, enzyme assays | ~0.5% miscarriage risk; results take longer (need to culture amniocytes) |
| Cordocentesis (PUBS) | ≥18 weeks | Direct fetal blood sampling from umbilical vein | Highest risk (~2%); rapid karyotype; used for fetal blood disorders |
| Cell-free fetal DNA (NIPT) | ≥10 weeks | Screening for trisomies 21, 18, 13; sex chromosome abnormalities | Non-invasive (maternal blood); high sensitivity but still a screening test (positive results need confirmation) |
27 High-Yield Review & Board Pearls
| Concept | Key Point |
|---|---|
| DNA replication | Semiconservative; leading strand continuous, lagging strand discontinuous (Okazaki fragments) |
| Transcription | RNA Pol II makes mRNA; α-amanitin inhibits Pol II; 5′ cap + poly-A tail + splicing |
| Translation | 30S: aminoglycosides, tetracyclines; 50S: macrolides, chloramphenicol, clindamycin, linezolid |
| Point mutations | Silent, missense (sickle cell = Glu→Val), nonsense (premature stop); frameshift (Duchenne) |
| DNA repair | NER → xeroderma pigmentosum; MMR → Lynch syndrome; HR → BRCA cancers |
| AD inheritance | 50% risk; structural proteins; variable expressivity; NF1, Marfan, Huntington, ADPKD |
| AR inheritance | 25% risk; enzyme deficiencies; carrier risk 2/3 for unaffected sibling; CF, sickle cell, PKU |
| XLR inheritance | Males affected; no male-to-male; Duchenne, hemophilia A/B, G6PD, Fabry |
| Trinucleotide repeats | Anticipation; Huntington (CAG), fragile X (CGG), myotonic dystrophy (CTG), Friedreich (GAA) |
| Imprinting | Prader–Willi = loss of paternal 15q; Angelman = loss of maternal 15q (UBE3A) |
| Trisomy 21 | Flat facies, ASD/VSD, duodenal atresia, ALL, early Alzheimer; 95% nondisjunction |
| Turner (45,X) | Short female, streak gonads, coarctation, webbed neck, no Barr body |
| Klinefelter (47,XXY) | Tall male, small testes, gynecomastia, infertility, one Barr body |
| Tumor suppressors | Two-hit hypothesis; p53 (Li–Fraumeni), Rb (retinoblastoma), APC (FAP), BRCA1/2 |
| Hardy–Weinberg | q² = disease prevalence; carrier frequency = 2pq; p + q = 1 |
| Genetic testing | CMA first-line for ID/MCA; FISH for known microdeletions; NGS for undiagnosed conditions |
| Pharmacogenomics | HLA-B*5701 before abacavir; CYP2D6 for codeine; TPMT for thiopurines |
Classic Associations
| Finding | Think… |
|---|---|
| Cherry-red macula + neurodegeneration in infant | Tay–Sachs disease (hexosaminidase A deficiency) |
| Blue sclerae + fractures | Osteogenesis imperfecta (type I collagen defect) |
| Lens subluxation upward + tall + aortic root dilation | Marfan syndrome (fibrillin-1) |
| Lens subluxation downward + intellectual disability + thromboembolic events | Homocystinuria (cystathionine synthase deficiency) |
| Café-au-lait spots + neurofibromas + Lisch nodules | NF1 (neurofibromin) |
| Bilateral acoustic schwannomas | NF2 (merlin) |
| Ash-leaf spots + seizures + facial angiofibromas | Tuberous sclerosis |
| Hepatosplenomegaly + “crinkled paper” macrophages | Gaucher disease (glucocerebrosidase deficiency) |
| Recurrent infections + absent B cells + male infant >6 months | Bruton agammaglobulinemia (BTK deficiency) |
| Self-mutilation + hyperuricemia + gout in a child | Lesch–Nyhan syndrome (HGPRT deficiency) |
| Musty body odor + fair skin + intellectual disability | Phenylketonuria (PAH deficiency) |
| Kayser–Fleischer rings + liver disease + psychiatric symptoms | Wilson disease (ATP7B) |
Ethical & Legal Issues in Genetics
| Issue | Key Considerations |
|---|---|
| Informed consent | Patients must understand purpose, implications, limitations, and possible results before testing |
| Genetic discrimination | GINA (Genetic Information Nondiscrimination Act, 2008) prohibits discrimination in employment and health insurance based on genetic information; does not cover life, disability, or long-term care insurance |
| Presymptomatic testing | Testing for adult-onset conditions (Huntington) raises issues of autonomy, right not to know, and psychological impact; counseling before and after testing is essential |
| Duty to warn | Tension between patient confidentiality and obligation to inform at-risk relatives; varies by jurisdiction |
| Pediatric genetic testing | Testing for adult-onset conditions in minors is generally deferred unless intervention in childhood would change management |
| Incidental findings | Genomic sequencing may reveal variants unrelated to the indication; ACMG recommends reporting pathogenic variants in ~80 actionable genes |
| Direct-to-consumer testing | Limited clinical validity for many results; may cause anxiety without proper counseling; incomplete variant coverage |
Epigenetic Therapies in Cancer
- DNMT inhibitors (azacitidine, decitabine) — demethylate silenced tumor suppressor genes; used in MDS and AML
- HDAC inhibitors (vorinostat, romidepsin) — increase histone acetylation → re-express silenced genes; used in cutaneous T-cell lymphoma
- IDH inhibitors (ivosidenib, enasidenib) — block oncometabolite 2-hydroxyglutarate production; used in IDH-mutant AML
- EZH2 inhibitors (tazemetostat) — block H3K27 trimethylation; used in EZH2-mutant follicular lymphoma
Board Exam Strategies for Genetics Questions
- When given a pedigree, first determine the inheritance pattern, then calculate risk
- For enzyme deficiency questions, think autosomal recessive unless stated otherwise
- For structural protein defects, think autosomal dominant
- If a question mentions consanguinity, suspect an AR disorder
- If only males are affected across generations (through carrier mothers), think X-linked recessive
- For trinucleotide repeat questions, identify the repeat, the gene, and whether it shows anticipation through the maternal or paternal line
- For cancer genetics, know the difference between oncogenes (gain of function, one hit) and tumor suppressors (loss of function, two hits)
- For Hardy–Weinberg, start with q² = disease prevalence and solve from there