Immunology
Innate and adaptive immunity, complement, antibodies, T-cell and B-cell development, hypersensitivity, immunodeficiency, autoimmunity, transplant immunology, and every immune mechanism, receptor, and clinical application across the full scope of medical immunology.
01 Overview & Significance
Immunology is the study of the immune system — the network of cells, tissues, organs, and soluble mediators that defend the body against infection and aberrant cells while maintaining tolerance to self. It integrates molecular biology, cell biology, genetics, and clinical medicine, and underpins understanding of infectious disease, autoimmunity, allergy, cancer, transplantation, and immunodeficiency. On USMLE Step 1, immunology accounts for approximately 9–15% of questions, making it one of the highest-yield foundational disciplines.
Every physician encounters immunologic disease daily — from anaphylaxis to autoimmune disorders to organ rejection. A solid immunologic foundation enables you to understand why vaccines work, why transplant patients need immunosuppression, why HIV destroys immunity, and how checkpoint inhibitors fight cancer. Immunology is the key that unlocks rational clinical reasoning across every specialty.
Scope of Medical Immunology
| Domain | Key Topics |
|---|---|
| Innate Immunity | Barriers, complement, phagocytes, pattern recognition receptors, NK cells |
| Adaptive Immunity | T cells, B cells, antibodies, MHC, antigen processing, immunologic memory |
| Hypersensitivity | Types I–IV reactions, allergy, anaphylaxis, serum sickness |
| Immunodeficiency | Primary (SCID, Bruton, DiGeorge) and acquired (HIV/AIDS) |
| Autoimmunity | SLE, RA, type 1 DM, Hashimoto, Graves, MS, autoantibodies |
| Transplant Immunology | Rejection types, HLA matching, GvHD, immunosuppressant drugs |
| Tumor Immunology | Immune surveillance, checkpoint inhibitors, CAR-T therapy |
| Vaccination | Live vs. inactivated, adjuvants, herd immunity, mRNA vaccines |
Historical Milestones
| Year | Milestone | Significance |
|---|---|---|
| 1796 | Jenner — cowpox vaccination | First deliberate immunization; foundation of vaccinology |
| 1880s | Pasteur — attenuated vaccines (anthrax, rabies) | Demonstrated that weakened pathogens confer immunity |
| 1890 | von Behring & Kitasato — antitoxin therapy | Discovery of humoral immunity (serum-mediated protection) |
| 1901 | Landsteiner — ABO blood groups | Basis of transfusion medicine and understanding of alloimmunity |
| 1953 | Grubb, Grubb & Grubb — agammaglobulinemia described | First primary immunodeficiency characterized |
| 1957 | Burnet — clonal selection theory | Each lymphocyte bears a unique receptor; antigen selects responding clone |
| 1975 | Köhler & Milstein — monoclonal antibody technology | Hybridoma technology revolutionized diagnostics and therapeutics |
| 2018 | Allison & Honjo — Nobel Prize for checkpoint inhibitors | CTLA-4 and PD-1 blockade transformed cancer immunotherapy |
02 Core Principles & Key Terminology
Fundamental Organizing Concepts
| Concept | Definition |
|---|---|
| Innate immunity | Rapid, non-specific, no memory; present at birth; uses germline-encoded receptors |
| Adaptive immunity | Slow onset (days), highly specific, generates memory; requires antigen exposure; uses somatically recombined receptors (TCR, BCR) |
| Humoral immunity | B-cell/antibody-mediated; defends against extracellular pathogens and toxins |
| Cell-mediated immunity | T-cell-mediated; defends against intracellular pathogens, tumors, transplants |
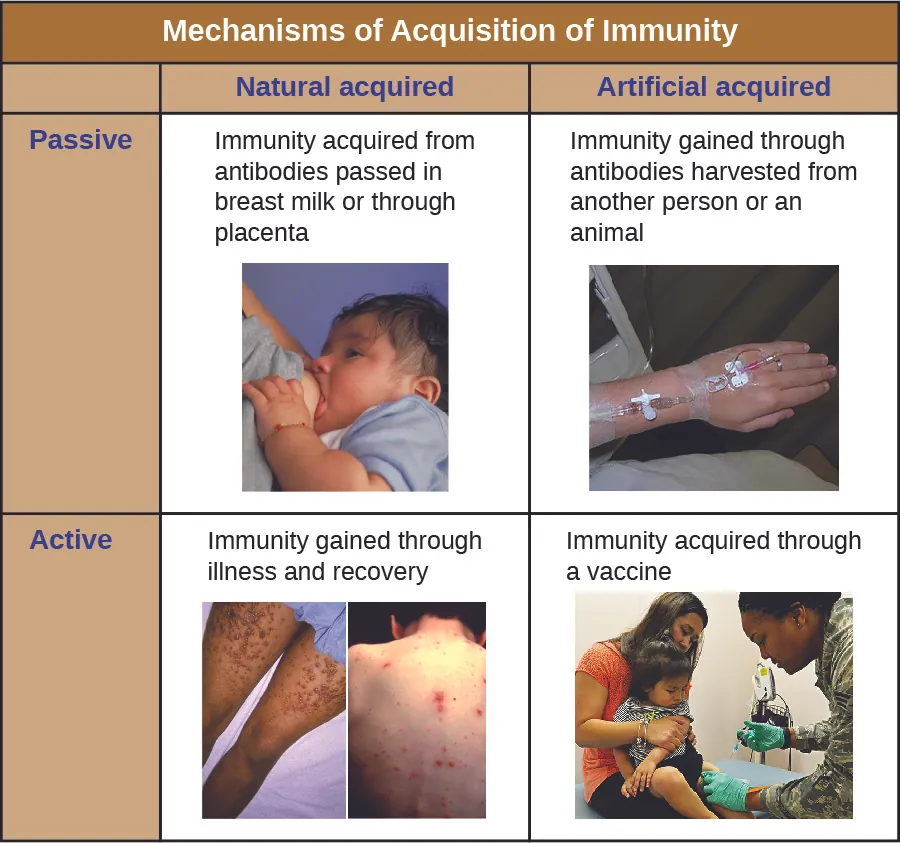
| Active immunity | Host generates own immune response (natural infection or vaccination); long-lasting |
| Passive immunity | Preformed antibodies transferred (maternal IgG, IVIg, antiserum); immediate but transient |
| Antigen | Any molecule recognized by the adaptive immune system; epitope = the specific region bound by antibody or TCR |
| Immunogen | An antigen capable of eliciting an immune response (all immunogens are antigens, but not all antigens are immunogens — e.g., haptens) |
| Hapten | Small molecule that is antigenic but not immunogenic alone; must bind a carrier protein to elicit a response (e.g., penicillin, poison ivy urushiol) |
| Adjuvant | Substance that enhances the immune response to a co-administered antigen (e.g., alum, MF59) |
Key Abbreviations
| Abbreviation | Meaning |
|---|---|
| APC | Antigen-presenting cell (dendritic cell, macrophage, B cell) |
| TCR | T-cell receptor (recognizes peptide–MHC complex) |
| BCR | B-cell receptor (membrane-bound immunoglobulin) |
| MHC | Major histocompatibility complex (HLA in humans) |
| Ig | Immunoglobulin (IgG, IgA, IgM, IgE, IgD) |
| PAMP | Pathogen-associated molecular pattern |
| PRR | Pattern recognition receptor (TLR, NLR, RLR, CLR) |
| MAC | Membrane attack complex (C5b–C9) |
| DAMP | Damage-associated molecular pattern (released from injured host cells) |
Innate vs. Adaptive Immunity: Detailed Comparison
| Feature | Innate Immunity | Adaptive Immunity |
|---|---|---|
| Onset | Immediate (minutes–hours) | Delayed (days–weeks on first exposure) |
| Specificity | Broad (recognizes general molecular patterns) | Highly specific (recognizes unique epitopes) |
| Memory | No classical memory (some “trained immunity” in monocytes/NK cells) | Robust immunologic memory (faster, stronger on re-exposure) |
| Receptors | Germline-encoded, limited diversity (PRRs: TLRs, NLRs, CLRs) | Somatically recombined, vast diversity (TCR: ~1015; BCR: ~1011) |
| Key cells | Neutrophils, macrophages, dendritic cells, NK cells, mast cells, eosinophils, basophils | T cells (CD4+, CD8+), B cells, plasma cells |
| Soluble mediators | Complement, cytokines, antimicrobial peptides, acute-phase proteins | Antibodies, cytokines |
| Self/non-self discrimination | Recognizes PAMPs (absent in host); DAMPs (host damage signals) | Clonal selection + central/peripheral tolerance mechanisms |
| Germ-line encoded? | Yes (fixed receptor repertoire) | No (generated by VDJ recombination; RAG-1/RAG-2 dependent) |
03 Cells of the Immune System
Myeloid Lineage
| Cell | Key Markers | Function |
|---|---|---|
| Neutrophil | CD66b; multilobed nucleus | First responder; phagocytosis; NETs (neutrophil extracellular traps); most abundant WBC (60–70%) |
| Monocyte / Macrophage | CD14, CD16; kidney-shaped nucleus | Phagocytosis, antigen presentation (MHC II), cytokine secretion; tissue-specific names (Kupffer cells in liver, microglia in CNS, alveolar macrophages in lung) |
| Dendritic cell | CD11c, MHC II (high) | Most potent APC; bridges innate and adaptive immunity; captures antigen in tissues, migrates to lymph nodes to activate naïve T cells |
| Eosinophil | CD193 (CCR3); bilobed nucleus | Defense against helminths (parasites); major basic protein release; role in allergic inflammation |
| Basophil | CD123; bilobed, S-shaped nucleus | IgE-mediated reactions; release histamine and heparin; circulating counterpart of mast cells |
| Mast cell | CD117 (c-Kit), FcεRI | Tissue-resident; IgE cross-linking triggers degranulation (histamine, tryptase, leukotrienes); central to Type I hypersensitivity |
Lymphoid Lineage
| Cell | Key Markers | Function |
|---|---|---|
| CD4+ T cell (helper) | CD3, CD4, TCRαβ | Orchestrates immune responses; recognizes MHC II–peptide; differentiates into Th1, Th2, Th17, Treg, Tfh |
| CD8+ T cell (cytotoxic) | CD3, CD8, TCRαβ | Kills virus-infected and tumor cells; recognizes MHC I–peptide; releases perforin and granzymes |
| B cell | CD19, CD20, CD21, surface Ig | Produces antibodies; antigen presentation to T cells; differentiates into plasma cells and memory B cells |
| Plasma cell | CD38, CD138; eccentric nucleus, clock-face chromatin | Terminally differentiated B cell; antibody factory (secretes ~2,000 Ab molecules/second) |
| NK cell | CD16, CD56; no TCR | Innate lymphocyte; kills cells lacking MHC I (missing self); ADCC via FcγRIII (CD16); releases perforin and granzymes |
| γδ T cell | CD3, TCRγδ | Bridge innate/adaptive; recognize non-peptide antigens (phospholipids, lipids); found in epithelial surfaces |
CD4 count in HIV: Normal >500 cells/μL. AIDS defined as <200 cells/μL. Opportunistic infections correlate with CD4 thresholds: <200 = PCP, <100 = Toxoplasma/Cryptococcus, <50 = MAC/CMV.
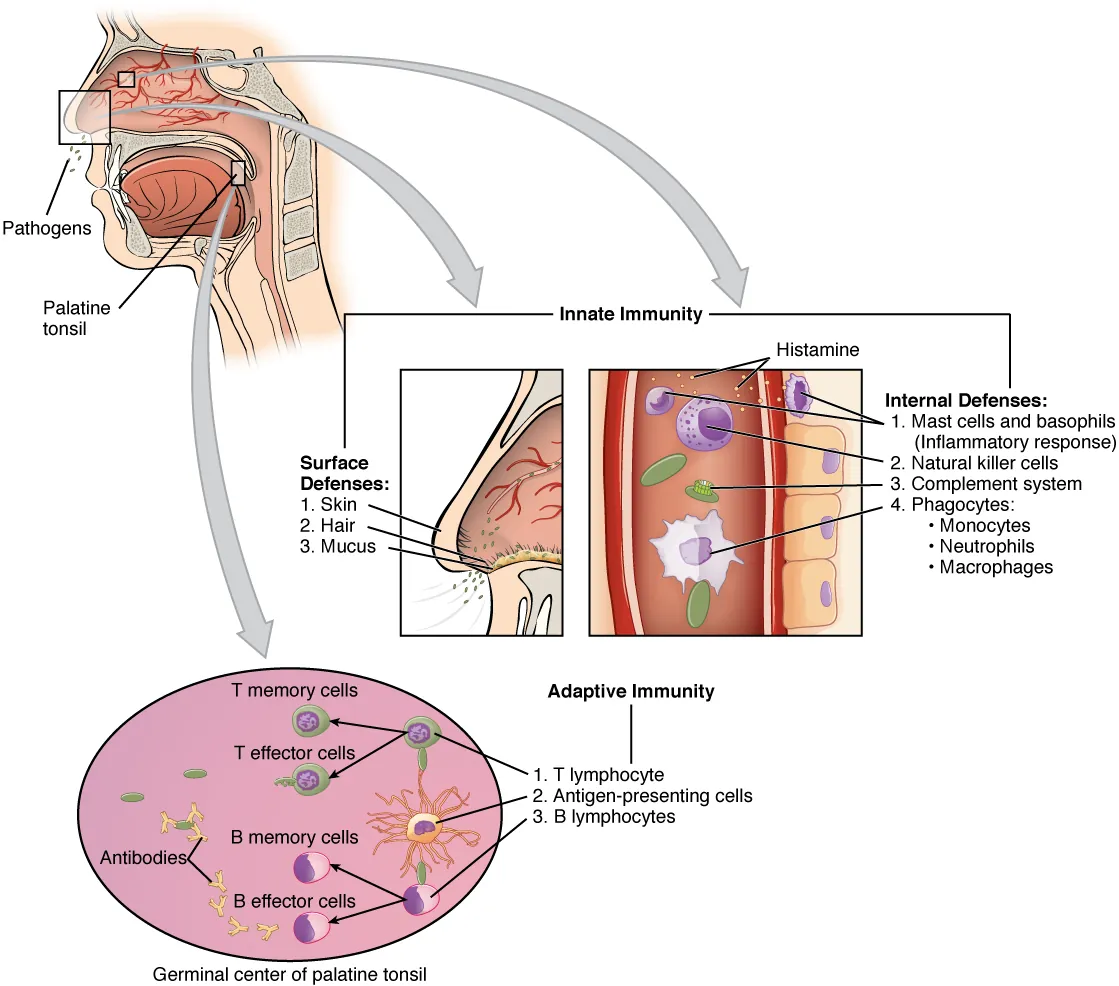
04 Barriers & Physical Defenses
The first line of defense consists of physical, chemical, and biological barriers that prevent pathogen entry without requiring immune cell activation.
| Barrier Type | Mechanism | Examples |
|---|---|---|
| Mechanical | Physical exclusion of pathogens | Skin (stratified squamous epithelium), mucociliary escalator, urinary flow, peristalsis, cough reflex |
| Chemical | Antimicrobial molecules | Stomach acid (pH 1–2), lysozyme (tears, saliva), defensins (skin, GI), lactoferrin (sequester iron), surfactant proteins A & D (lung) |
| Biological | Commensal flora competition | Normal flora of skin, gut, vagina compete with pathogens for nutrients and produce bacteriocins; disruption leads to opportunistic infections (e.g., C. difficile after antibiotics) |
Antimicrobial Peptides
Antimicrobial peptides (AMPs) are small cationic molecules that insert into microbial membranes, disrupting their integrity. Key families include:
| Peptide | Location | Function |
|---|---|---|
| Defensins (α and β) | α-defensins: neutrophil granules, Paneth cells (small intestine); β-defensins: epithelial surfaces (skin, respiratory, GI) | Form pores in microbial membranes; chemotactic for dendritic cells and T cells |
| Cathelicidin (LL-37) | Neutrophils, keratinocytes, epithelial cells | Antimicrobial; enhances TLR signaling; vitamin D upregulates expression (link between vitamin D deficiency and infection susceptibility) |
| Lysozyme | Tears, saliva, nasal secretions, neutrophil granules | Cleaves peptidoglycan in bacterial cell walls (more effective against Gram-positives) |
| Lactoferrin | Mucosal secretions, neutrophil granules, breast milk | Sequesters iron from bacteria (iron-dependent organisms); direct bactericidal activity |
05 Pattern Recognition & Toll-Like Receptors
Innate immune cells detect pathogens through pattern recognition receptors (PRRs) that recognize conserved microbial structures called pathogen-associated molecular patterns (PAMPs) and host-derived danger signals called damage-associated molecular patterns (DAMPs).
Major PRR Families
| PRR Family | Location | Ligands | Downstream Effect |
|---|---|---|---|
| TLRs (Toll-like receptors) | Cell surface (1,2,4,5,6) or endosome (3,7,8,9) | LPS, flagellin, dsRNA, ssRNA, CpG DNA | NF-κB activation → pro-inflammatory cytokines (TNF-α, IL-1, IL-6) |
| NLRs (NOD-like receptors) | Cytoplasm | Peptidoglycan (NOD1/2), DAMPs (NLRP3 inflammasome) | Inflammasome assembly → caspase-1 → IL-1β and IL-18 secretion |
| RLRs (RIG-I-like receptors) | Cytoplasm | Viral dsRNA | Type I interferon (IFN-α/β) production |
| CLRs (C-type lectin receptors) | Cell surface | Mannose, β-glucan (fungi) | Phagocytosis, cytokine production |
| cGAS-STING | Cytoplasm | Cytosolic dsDNA | Type I interferon production |
Key Toll-Like Receptors
| TLR | PAMP Recognized | Pathogen |
|---|---|---|
| TLR-1/2 | Bacterial lipopeptides | Gram-positive bacteria |
| TLR-3 | dsRNA | Viruses (endosomal) |
| TLR-4 | LPS (lipopolysaccharide) | Gram-negative bacteria |
| TLR-5 | Flagellin | Flagellated bacteria |
| TLR-7/8 | ssRNA | Viruses (endosomal) |
| TLR-9 | Unmethylated CpG DNA | Bacteria, DNA viruses (endosomal) |
The NLRP3 inflammasome is activated by diverse danger signals (uric acid crystals in gout, cholesterol crystals in atherosclerosis, asbestos, silica). Assembly activates caspase-1, which cleaves pro-IL-1β and pro-IL-18 into active forms and triggers pyroptosis (inflammatory cell death via gasdermin D pores). Mutations causing constitutive NLRP3 activation cause cryopyrin-associated periodic syndromes (CAPS), treated with IL-1 antagonists (anakinra, canakinumab).
06 Complement System
The complement system is a cascade of >30 plasma proteins that enhance (“complement”) the ability of antibodies and phagocytes to clear pathogens. Its three main functions are opsonization (C3b), inflammation (C3a, C5a — anaphylatoxins), and lysis (membrane attack complex, C5b–C9).
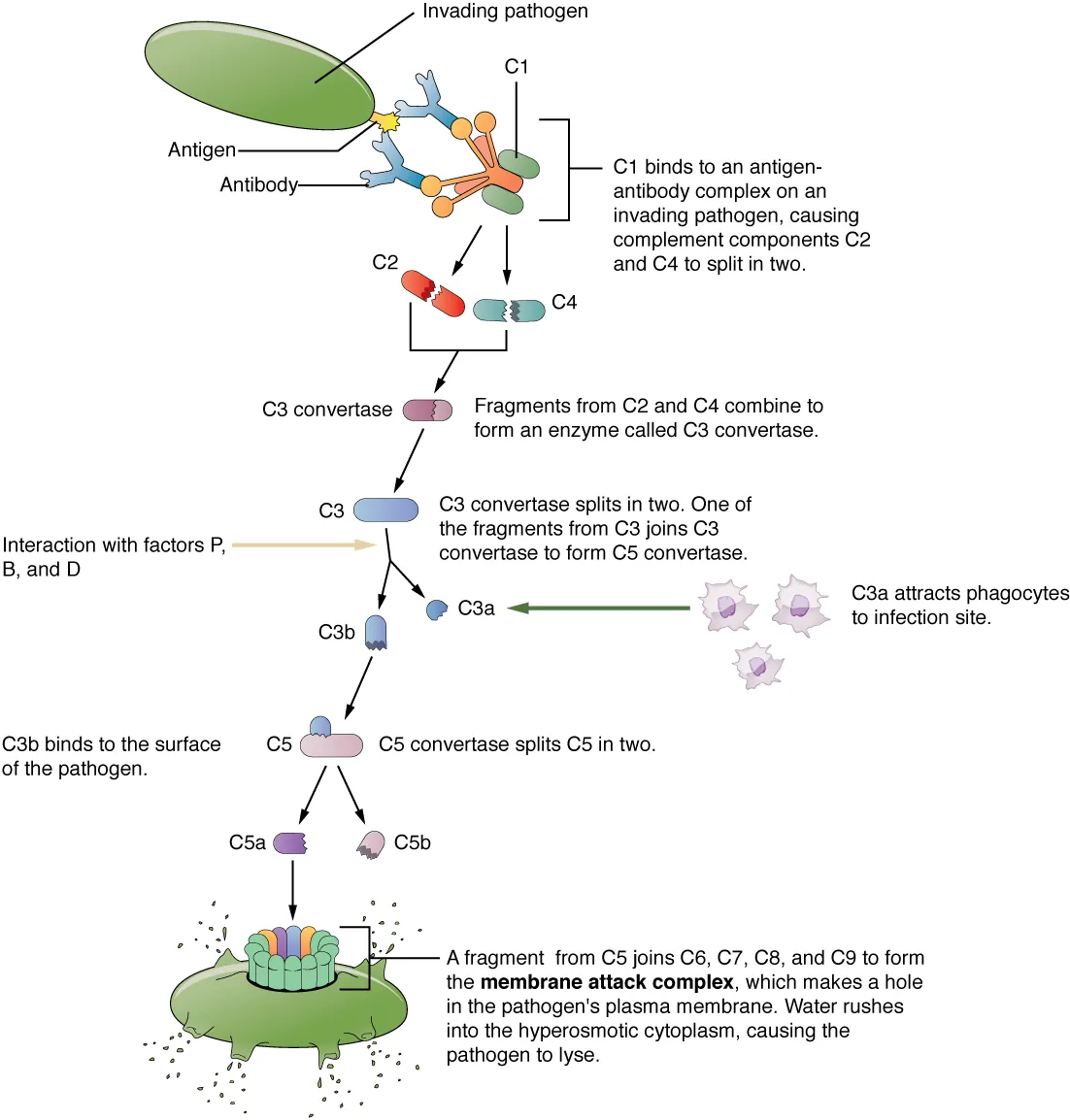
Activation Pathways
| Pathway | Trigger | Key Components | C3 Convertase |
|---|---|---|---|
| Classical | IgG or IgM–antigen complexes bind C1q | C1q, C1r, C1s, C4, C2 | C4b2a |
| Lectin (MBL) | Mannose-binding lectin binds mannose on pathogens | MBL, MASP-1/2, C4, C2 | C4b2a |
| Alternative | Spontaneous C3 hydrolysis (tick-over) on pathogen surfaces | C3, Factor B, Factor D, Properdin | C3bBb |
All three pathways converge at C3 convertase, which cleaves C3 into C3a (anaphylatoxin) and C3b (opsonin). C3b joins the C3 convertase to form C5 convertase, which cleaves C5 into C5a (most potent anaphylatoxin and neutrophil chemoattractant) and C5b, initiating the membrane attack complex (MAC) (C5b–C6–C7–C8–C9).
Complement Functions & Key Fragments
| Fragment | Function |
|---|---|
| C3b | Opsonization (binds pathogen surface, recognized by CR1 on phagocytes) |
| C3a, C4a, C5a | Anaphylatoxins — trigger mast cell degranulation, smooth muscle contraction, vascular permeability; C5a is the most potent |
| C5a | Neutrophil chemotaxis (strongest complement-derived chemoattractant) |
| C5b–C9 (MAC) | Forms pores in cell membranes → osmotic lysis (especially effective against Neisseria) |
Complement Regulatory Proteins
| Regulator | Mechanism | Clinical Relevance |
|---|---|---|
| Decay-accelerating factor (DAF/CD55) | Accelerates decay of C3/C5 convertases | Deficiency of DAF and CD59 (GPI-anchored) → Paroxysmal nocturnal hemoglobinuria (PNH) |
| CD59 (protectin) | Prevents MAC assembly on host cells | |
| C1 esterase inhibitor (C1-INH) | Inhibits C1r/C1s (classical pathway) | Deficiency → Hereditary angioedema (excess bradykinin from unchecked complement/kinin activation) |
| Factor H | Cofactor for Factor I–mediated cleavage of C3b | Mutations → atypical hemolytic uremic syndrome (aHUS) |
C1–C4 deficiency → SLE-like syndrome (impaired immune complex clearance). C3 deficiency → severe recurrent pyogenic infections (no opsonization). C5–C9 (MAC) deficiency → recurrent Neisseria infections (meningococcal meningitis, gonococcal disease). C1-INH deficiency → hereditary angioedema. DAF/CD59 deficiency → PNH (complement-mediated hemolysis).
Complement in Disease & Therapy
| Clinical Scenario | Complement Finding | Mechanism |
|---|---|---|
| Active SLE / PSGN / cryoglobulinemia | Low C3 and C4 (consumed) | Immune complex activation of classical pathway consumes complement |
| Hereditary angioedema | Low C4, low C1-INH (functional or quantitative) | Uninhibited C1 cleaves C4; bradykinin excess causes angioedema |
| Alternative pathway activation (e.g., aHUS, C3 GN) | Low C3, normal C4 | Alternative pathway preferentially consumes C3 without affecting C4 |
| PNH | Normal complement levels; RBCs hypersensitive to complement lysis | GPI anchor deficiency → absent DAF/CD59 on RBCs |
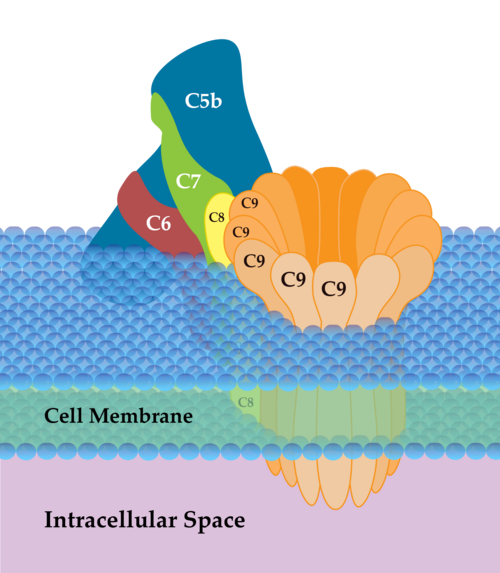
Eculizumab (anti-C5 monoclonal antibody) blocks C5 cleavage, preventing MAC formation and C5a generation. It is used in PNH, atypical HUS, and myasthenia gravis. Patients on eculizumab must be vaccinated against Neisseria meningitidis (meningococcal vaccine) because blocking the MAC increases susceptibility to meningococcal disease.
07 Phagocytosis & Inflammation
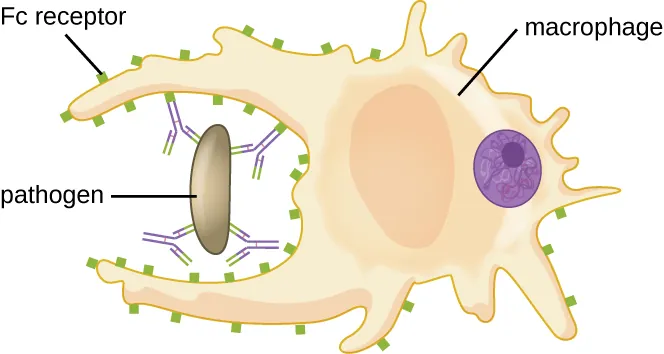
Steps of Phagocytosis
Phagocytosis follows an orderly sequence: (1) Chemotaxis — neutrophils/macrophages migrate toward chemoattractants (C5a, IL-8/CXCL8, LTB4, bacterial peptides like fMLP); (2) Opsonization — pathogen coating with IgG (Fc receptor binding) or C3b (CR1 binding); (3) Engulfment — pseudopod extension and phagosome formation; (4) Killing — phagosome fuses with lysosome, activating oxygen-dependent and oxygen-independent mechanisms.
Microbial Killing Mechanisms
| Mechanism | Key Mediators | Clinical Relevance |
|---|---|---|
| Oxidative burst (O2-dependent) | NADPH oxidase → superoxide (O2−) → H2O2 → HOCl (bleach, via myeloperoxidase) | Deficient in Chronic Granulomatous Disease (CGD) — NADPH oxidase defect; diagnosed by negative nitroblue tetrazolium (NBT) or dihydrorhodamine (DHR) test |
| O2-independent | Lysozyme, lactoferrin, defensins, cathepsins, major basic protein (eosinophils) | Provide baseline killing even in anaerobic conditions |
| NETs (neutrophil extracellular traps) | Chromatin fibers studded with antimicrobial proteins extruded by neutrophils | Trap and kill extracellular pathogens; excessive NETosis implicated in SLE, vasculitis, thrombosis |
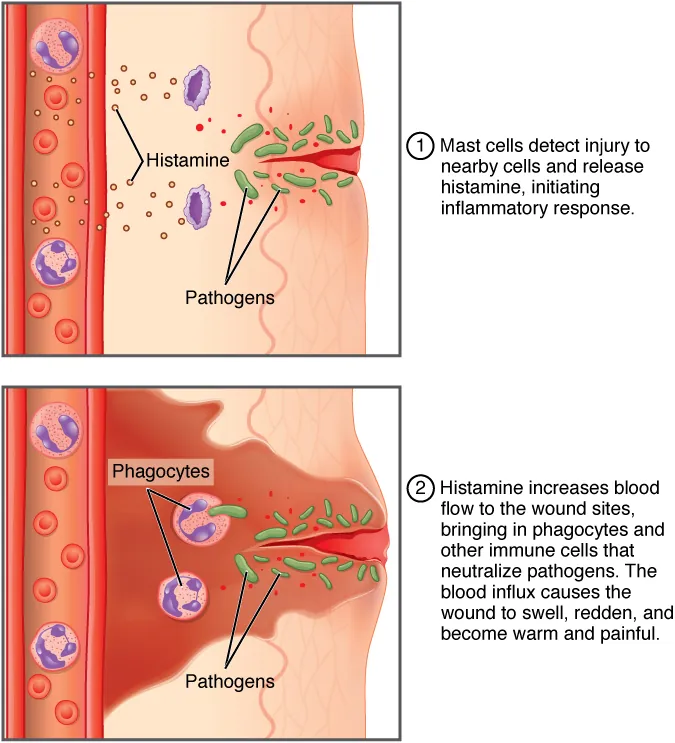
Cardinal Signs of Inflammation
Rubor (redness), calor (heat), tumor (swelling), dolor (pain), and functio laesa (loss of function). These result from vasodilation (histamine, PGE2, NO), increased vascular permeability (histamine, C3a/C5a, leukotrienes), and leukocyte infiltration.
Key Inflammatory Mediators
| Mediator | Source | Action |
|---|---|---|
| Histamine | Mast cells, basophils | Vasodilation, increased vascular permeability, bronchospasm |
| TNF-α | Macrophages | Fever, acute-phase proteins, endothelial activation, cachexia; dominant mediator in sepsis |
| IL-1 | Macrophages | Fever (acts on hypothalamus), endothelial activation, acute-phase protein synthesis |
| IL-6 | Macrophages, T cells | Fever, hepatic acute-phase protein synthesis (CRP, fibrinogen), B-cell differentiation |
| PGE2 | COX-2 in many cell types | Vasodilation, pain sensitization, fever; target of NSAIDs |
| LTB4 | Lipoxygenase pathway | Potent neutrophil chemoattractant |
| LTC4/D4/E4 | Mast cells, eosinophils | Bronchospasm, vasoconstriction, mucus secretion (slow-reacting substances of anaphylaxis) |
Leukocyte Extravasation (Diapedesis)
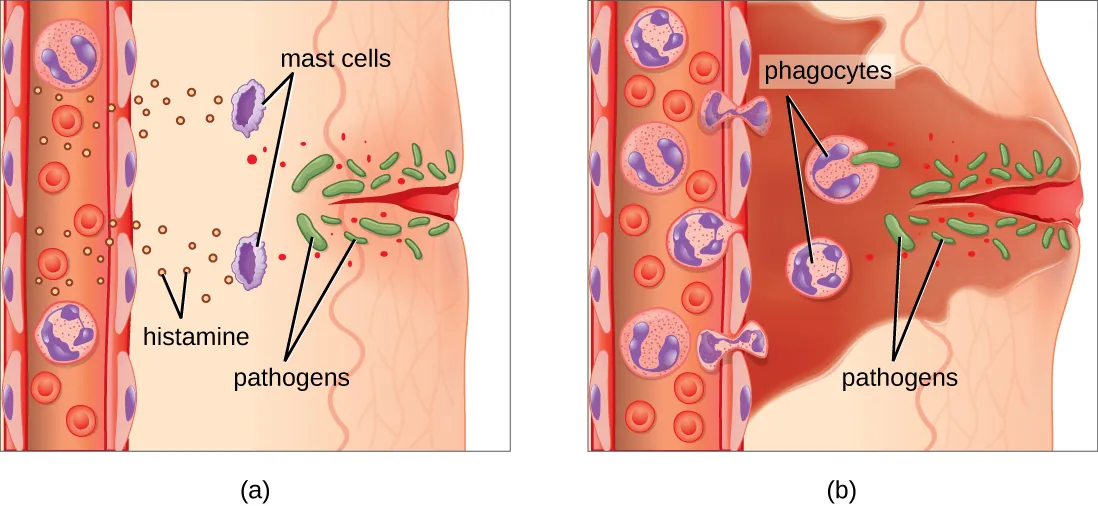
Leukocytes migrate from blood to tissues through a multi-step process at postcapillary venules:
| Step | Molecules Involved | Mechanism |
|---|---|---|
| 1. Rolling | E-selectin and P-selectin (endothelium) bind sialyl-LewisX (leukocyte) | Loose, reversible adhesion; leukocyte rolls along endothelium |
| 2. Activation | Chemokines (IL-8, C5a) activate leukocyte integrins | Conformational change in integrins increases binding affinity |
| 3. Firm adhesion | Integrins (LFA-1/CD11a-CD18) bind ICAM-1; VLA-4 binds VCAM-1 | Tight adhesion stops rolling; leukocyte flattens |
| 4. Transmigration | PECAM-1 (CD31) on both leukocyte and endothelium | Leukocyte squeezes between endothelial cells into tissue |
LAD Type 1: Deficiency of CD18 (β2-integrin subunit) prevents firm adhesion → neutrophils cannot leave bloodstream → leukocytosis, absent pus formation, delayed umbilical cord separation, recurrent bacterial infections. LAD Type 2: Defect in fucose metabolism (sialyl-LewisX absent) → defective rolling → similar but milder phenotype.
08 T-Cell Development & Thymic Selection
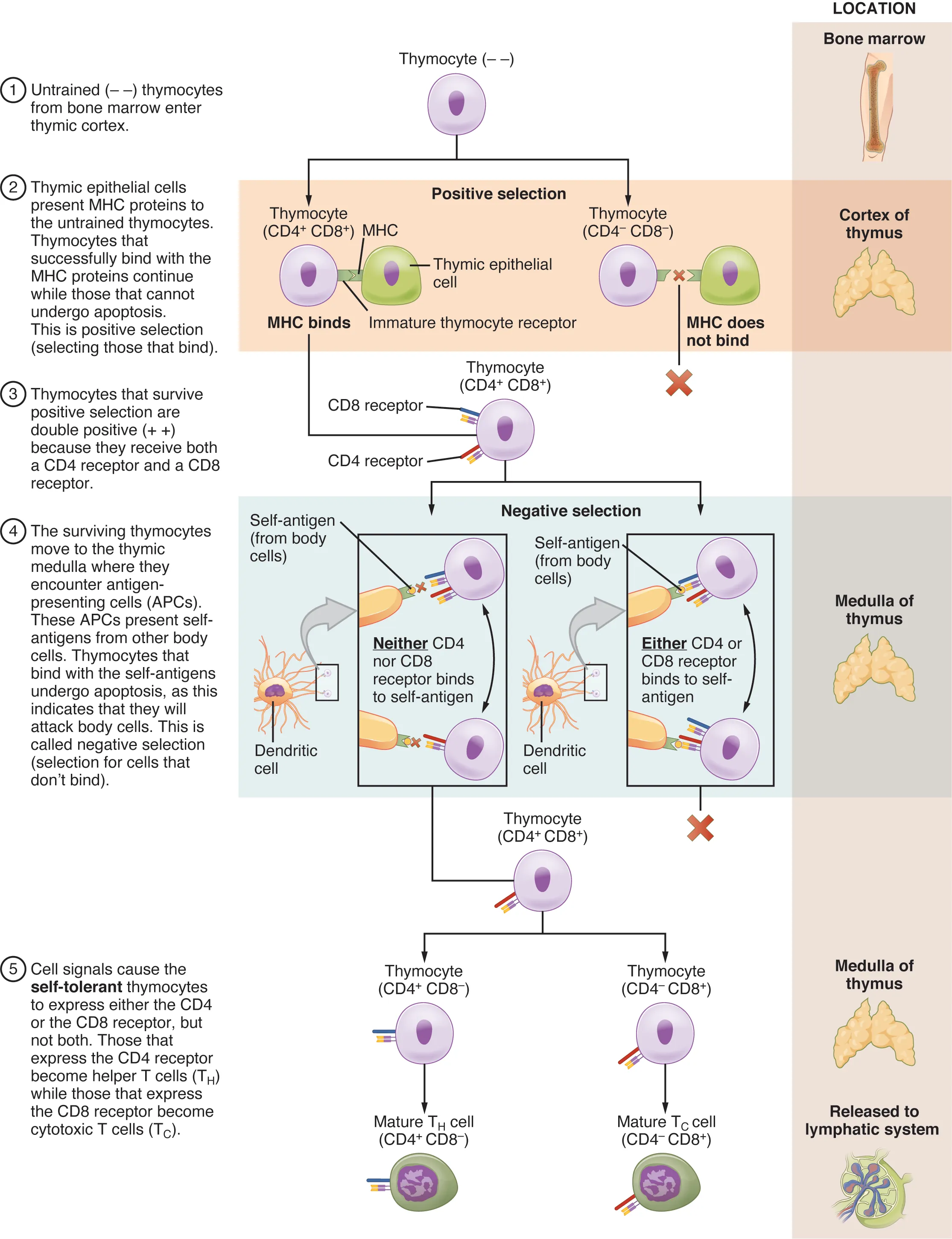
T-cell precursors originate from bone marrow hematopoietic stem cells and migrate to the thymus for maturation. The thymus is most active in childhood and undergoes progressive involution after puberty, though residual function persists into adulthood.
Stages of Thymic Development
| Stage | Phenotype | Location | Key Event |
|---|---|---|---|
| Double-negative (DN) | CD4− CD8− | Thymic cortex | TCR β-chain gene rearrangement (VDJ recombination via RAG-1/RAG-2); β-selection checkpoint |
| Double-positive (DP) | CD4+ CD8+ | Thymic cortex | TCR α-chain rearrangement; now express complete αβ-TCR |
| Positive selection | DP → SP | Thymic cortex | Cortical epithelial cells test for MHC recognition; T cells that bind MHC with moderate affinity survive; those that fail to bind → apoptosis (death by neglect) |
| Negative selection | SP | Cortico-medullary junction & medulla | Medullary epithelial cells and DCs present self-antigens (driven by AIRE gene); T cells that bind self-peptide–MHC too strongly → apoptosis (clonal deletion) or Treg differentiation |
| Single-positive (SP) | CD4+ or CD8+ | Thymic medulla | Mature, self-tolerant T cells exit to periphery |
The autoimmune regulator (AIRE) transcription factor enables thymic medullary epithelial cells to express tissue-restricted antigens (e.g., insulin, thyroglobulin) so that self-reactive T cells can be deleted. AIRE mutations cause Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy (APECED / APS-1) — characterized by chronic mucocutaneous candidiasis, hypoparathyroidism, and adrenal insufficiency.
Only about 2–5% of thymocytes survive both positive and negative selection. The rest die by apoptosis. This rigorous screening ensures that the T-cell repertoire is both self-MHC restricted (positive selection) and self-tolerant (negative selection).
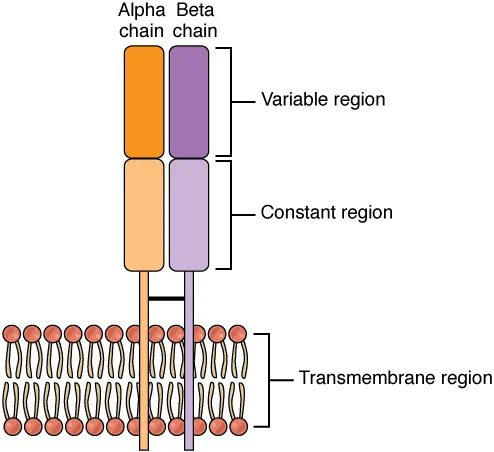
VDJ Recombination & TCR Diversity
The T-cell receptor (TCR) is generated through somatic recombination of variable (V), diversity (D), and joining (J) gene segments. This process, mediated by RAG-1 and RAG-2 recombinases, generates enormous receptor diversity from a limited number of germline gene segments.
| TCR Chain | Gene Segments | Recombination |
|---|---|---|
| α-chain | Vα and Jα segments | VJ recombination (similar to Ig light chain) |
| β-chain | Vβ, Dβ, and Jβ segments | VDJ recombination (D-J first, then V-DJ); β-selection checkpoint |
Additional diversity comes from junctional diversity (imprecise joining at segment boundaries) and N-nucleotide addition by terminal deoxynucleotidyl transferase (TdT). Unlike B cells, T cells do NOT undergo somatic hypermutation — TCR diversity is generated entirely during development. The estimated TCR repertoire is >1015 unique receptors.
09 MHC & Antigen Presentation
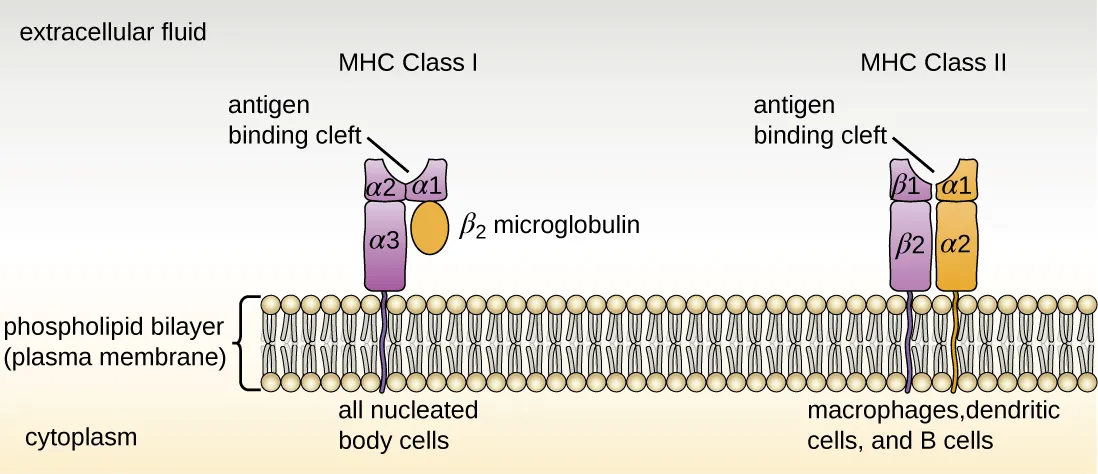
MHC Class I vs. Class II
| Feature | MHC Class I | MHC Class II |
|---|---|---|
| Genes (HLA) | HLA-A, HLA-B, HLA-C | HLA-DP, HLA-DQ, HLA-DR |
| Structure | α-chain + β2-microglobulin | α-chain + β-chain |
| Expression | All nucleated cells + platelets (not RBCs) | Professional APCs (dendritic cells, macrophages, B cells) |
| Peptide source | Endogenous (intracellular) — viral, tumor proteins | Exogenous (extracellular) — phagocytosed pathogens |
| Processing pathway | Proteasome → TAP transporter → ER loading | Endosome/lysosome → invariant chain (Ii) removed by cathepsin → CLIP replaced by peptide via HLA-DM |
| Recognized by | CD8+ cytotoxic T cells | CD4+ helper T cells |
| Binding groove | Closed (fits 8–10 aa peptides) | Open (fits 13–25 aa peptides) |
“MHC I loads from the Inside” (endogenous antigen from cytoplasm). “MHC II loads from the Outside” (exogenous antigen from endosomes). Rule of 8: MHC I × CD8 = 8. MHC II × CD4 = 8.
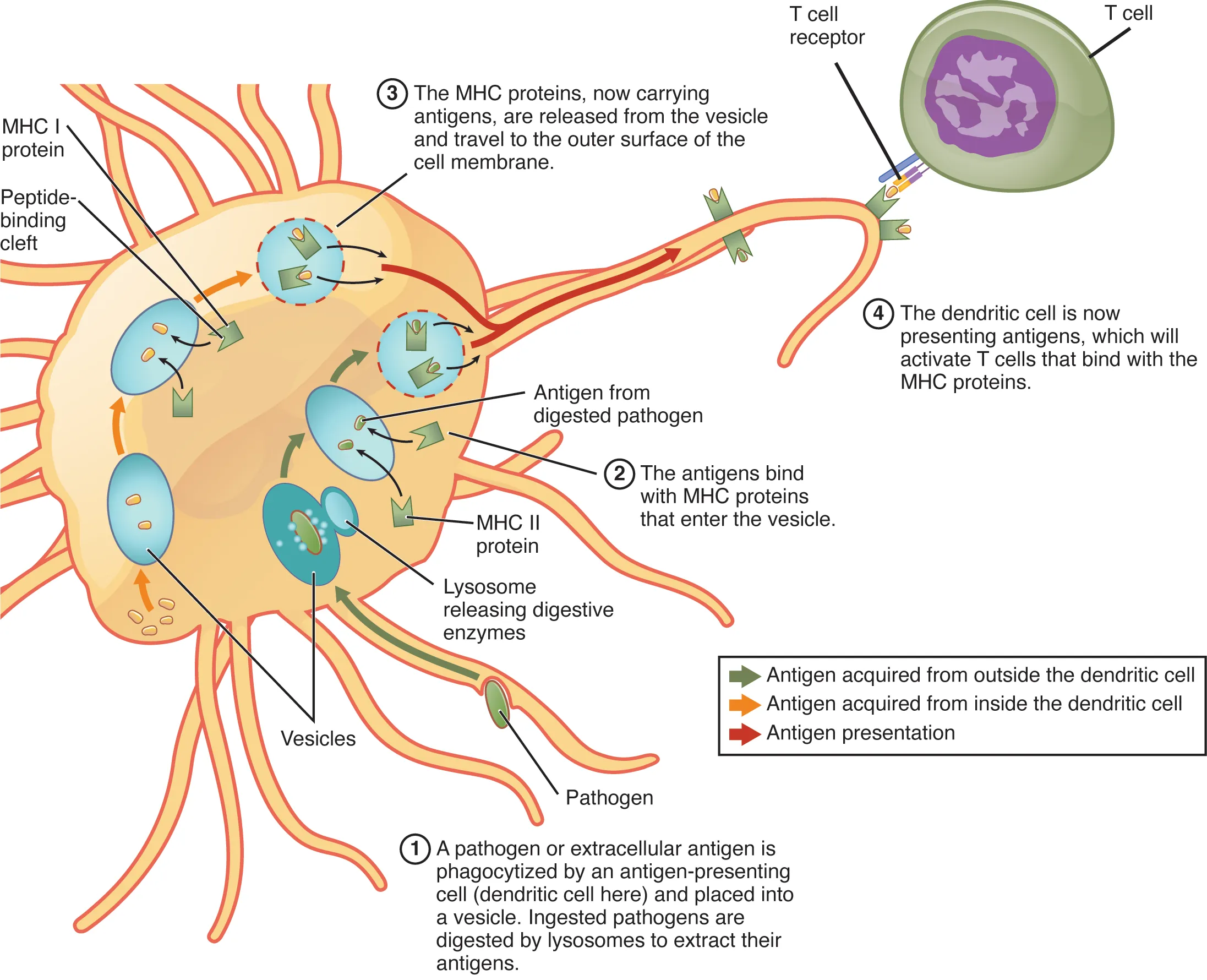
Antigen Processing Pathways in Detail
| Step | MHC I Pathway (Endogenous) | MHC II Pathway (Exogenous) |
|---|---|---|
| 1. Antigen source | Intracellular proteins (viral, tumor, self) | Extracellular proteins (phagocytosed, endocytosed) |
| 2. Processing | Proteasome degrades protein into 8–10 aa peptides | Acidic proteases in endosome/lysosome generate 13–25 aa peptides |
| 3. Transport to ER/loading | TAP transporter moves peptides into ER; peptide loaded onto MHC I–β2m complex with help of tapasin, calreticulin, ERp57 | MHC II assembled in ER with invariant chain (Ii/CD74) blocking groove; Ii degraded in endosome by cathepsin S, leaving CLIP fragment; HLA-DM catalyzes CLIP removal and peptide loading |
| 4. Surface display | MHC I–peptide transported to cell surface via Golgi | MHC II–peptide complex transported to cell surface |
| 5. T-cell recognition | CD8+ T cell (TCR + CD8 co-receptor) | CD4+ T cell (TCR + CD4 co-receptor) |
Cross-Presentation
Cross-presentation is the ability of certain dendritic cells to load exogenous antigens onto MHC I molecules, thereby activating CD8+ T cells against extracellular pathogens and tumors. This is essential for anti-tumor and anti-viral immunity when the DC itself is not infected. Defects in cross-presentation may contribute to immune evasion by tumors.
HLA Disease Associations
| HLA Allele | Associated Disease |
|---|---|
| HLA-B27 | Ankylosing spondylitis, reactive arthritis, psoriatic arthritis, IBD-associated arthritis |
| HLA-DR2 | Multiple sclerosis, SLE, Goodpasture syndrome |
| HLA-DR3 | Type 1 diabetes mellitus, SLE, Graves disease, celiac disease |
| HLA-DR4 | Type 1 diabetes mellitus, rheumatoid arthritis |
| HLA-DR5 | Hashimoto thyroiditis, pernicious anemia |
| HLA-DQ2/DQ8 | Celiac disease (DQ2 in ~95%, DQ8 in remainder) |
| HLA-B5801 | Allopurinol hypersensitivity (screen before prescribing in certain populations) |
Antigen Presentation Pathways — Special Scenarios
| Scenario | Mechanism | Clinical Significance |
|---|---|---|
| Cross-presentation | Exogenous antigen loaded onto MHC I (by specialized dendritic cells) | Essential for anti-tumor and anti-viral CTL responses when APC is not directly infected |
| Cross-dressing | Transfer of intact MHC–peptide complexes from donor cells to host APCs | Contributes to allograft rejection; donor MHC transferred to recipient DCs |
| Autophagy-mediated presentation | Intracellular antigens delivered to MHC II compartment via autophagy | Allows MHC II presentation of cytoplasmic antigens (e.g., EBV nuclear antigens) |
| CD1 presentation | CD1 molecules (non-classical MHC) present lipid antigens to NKT cells and γδ T cells | Important in mycobacterial immunity (mycolic acids, lipoarabinomannan) |
TAP Deficiency
TAP (transporter associated with antigen processing) is essential for loading peptides onto MHC I in the ER. TAP deficiency results in markedly reduced MHC I surface expression, leading to a clinical phenotype resembling bare lymphocyte syndrome type I: recurrent respiratory infections, bronchiectasis, and necrotizing granulomatous skin lesions. Importantly, CD8+ T cells are present but poorly functional due to inadequate MHC I–mediated positive selection.
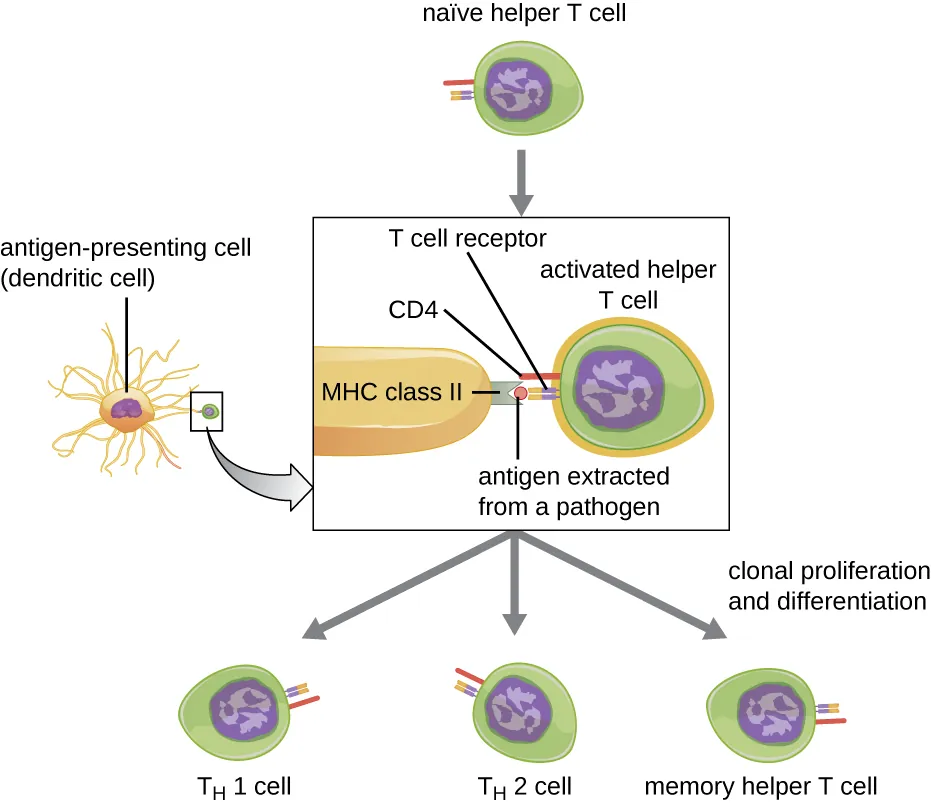
10 CD4+ T Helper Subsets (Th1, Th2, Th17, Treg)
Naïve CD4+ T cells differentiate into distinct effector subsets depending on the cytokine milieu at the time of activation. Each subset produces a characteristic cytokine profile and drives specific immune functions.
| Subset | Inducing Cytokines | Master Transcription Factor | Signature Cytokines | Function |
|---|---|---|---|---|
| Th1 | IL-12, IFN-γ | T-bet | IFN-γ, TNF-α, IL-2 | Activates macrophages (intracellular pathogens: mycobacteria, fungi); promotes cell-mediated immunity; class switch to IgG |
| Th2 | IL-4 | GATA-3 | IL-4, IL-5, IL-13, IL-10 | B-cell activation, class switch to IgE; eosinophil recruitment (IL-5); helminth defense; drives allergic responses |
| Th17 | IL-6, TGF-β, IL-23 | RORγt | IL-17, IL-22 | Neutrophil recruitment; mucosal immunity; defense against extracellular bacteria and fungi; implicated in autoimmunity (RA, MS, psoriasis) |
| Treg | TGF-β, IL-2 | FoxP3 | IL-10, TGF-β | Suppresses immune responses; maintains self-tolerance; prevents autoimmunity |
| Tfh (follicular helper) | IL-6, IL-21 | Bcl-6 | IL-21, IL-4 | Helps B cells in germinal centers; essential for affinity maturation, class switching, memory B-cell formation |
Th1 and Th2 responses are mutually inhibitory. IFN-γ (Th1) suppresses Th2 differentiation, while IL-4 and IL-10 (Th2) suppress Th1. An imbalanced Th2 response drives atopic disease (asthma, eczema, allergic rhinitis). An imbalanced Th1 response drives organ-specific autoimmunity. This balance is critical for understanding why some infections (e.g., lepromatous vs. tuberculoid leprosy) produce different clinical outcomes.
T-Cell Co-stimulation & Signal Integration
Full T-cell activation requires three signals:
| Signal | Interaction | Consequence of Absence |
|---|---|---|
| Signal 1 | TCR recognizes peptide–MHC complex | No activation (antigen not recognized) |
| Signal 2 | Co-stimulation: B7 (CD80/CD86) on APC binds CD28 on T cell | T-cell anergy (functional unresponsiveness) — basis of peripheral tolerance |
| Signal 3 | Cytokine milieu determines T-cell differentiation (IL-12 → Th1; IL-4 → Th2; TGF-β + IL-6 → Th17) | Default or unpolarized response |
Key co-stimulatory and co-inhibitory pairs:
- CD28–B7: Positive co-stimulation; essential for naïve T-cell activation
- CTLA-4–B7: Negative regulator; higher affinity than CD28; dampens activation (target of ipilimumab)
- PD-1–PD-L1/PD-L2: Negative regulator; induces T-cell exhaustion in periphery (target of pembrolizumab, nivolumab)
- CD40–CD40L: T-cell “licenses” APC; also critical for B-cell class switching (CD40L deficiency → Hyper-IgM syndrome)
- ICOS–ICOS-L: Important for Tfh function in germinal centers; ICOS deficiency causes CVID-like syndrome
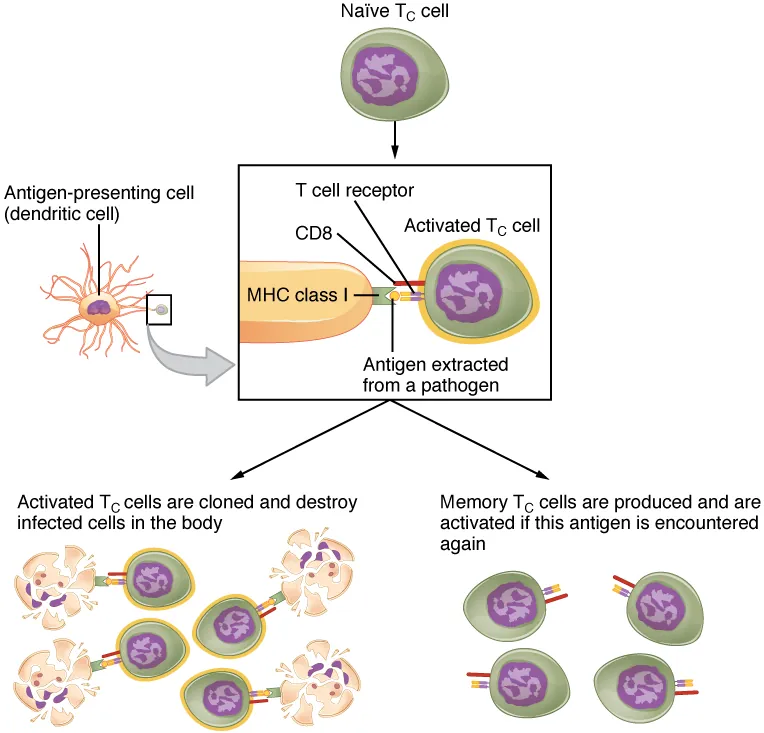
11 CD8+ Cytotoxic T Cells & NK Cells
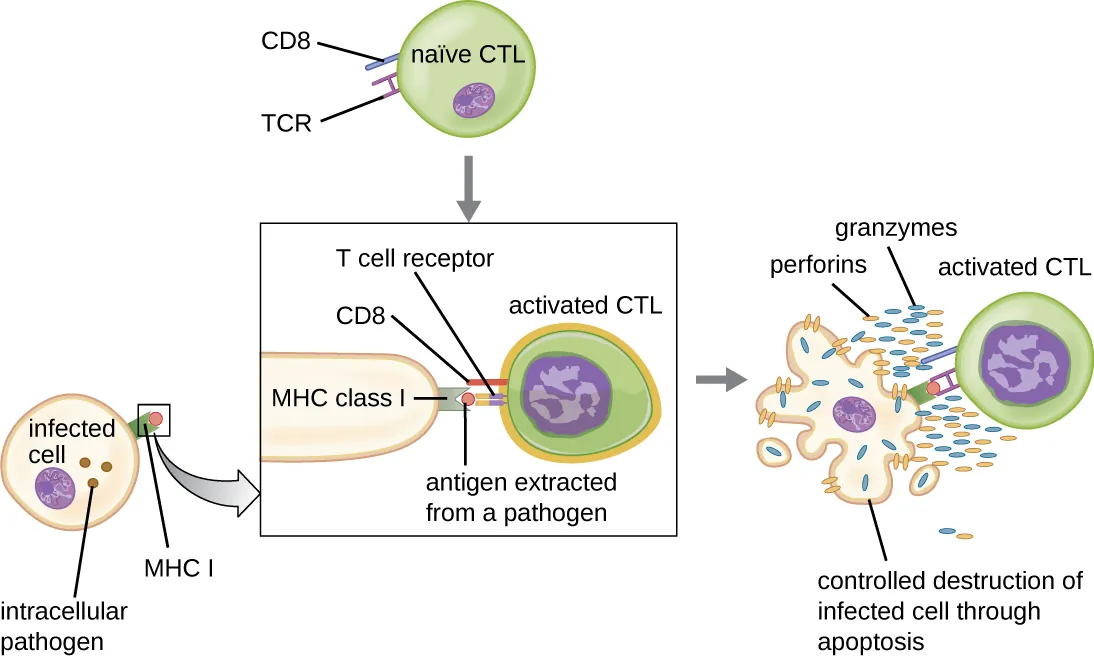
CD8+ Cytotoxic T Lymphocytes (CTLs)
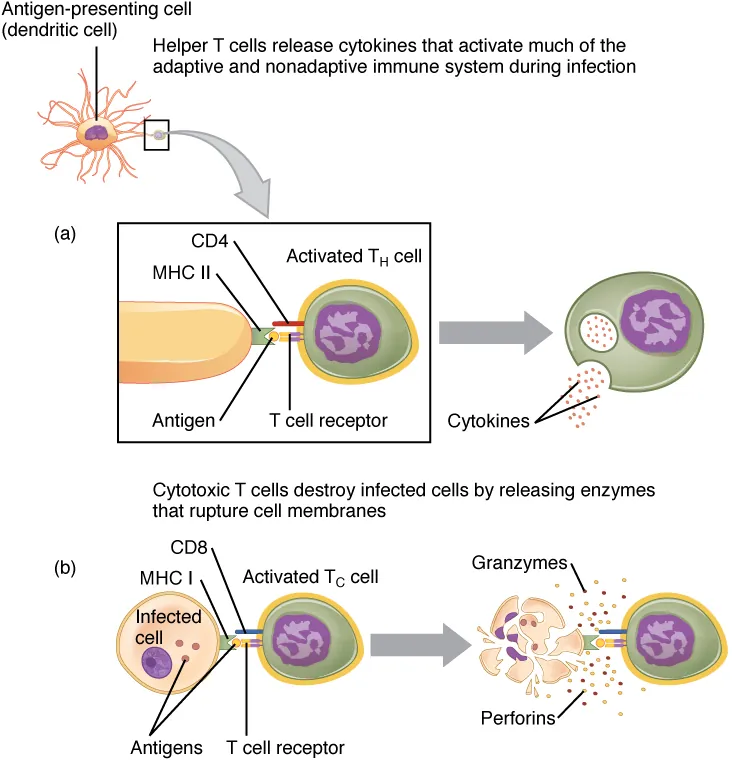
CD8+ T cells recognize endogenous peptides presented on MHC I. Upon activation (requiring signal 1 = TCR–MHC I, signal 2 = co-stimulation via B7–CD28, signal 3 = cytokines like IL-2), they differentiate into effector CTLs that kill target cells through two main mechanisms:
- Perforin/Granzyme pathway — Perforin creates pores in the target cell membrane; granzymes (serine proteases) enter and activate caspases, triggering apoptosis
- Fas/FasL pathway — FasL (CD95L) on CTL binds Fas (CD95) on target cell, activating caspase-8 and the extrinsic apoptosis pathway
Natural Killer (NK) Cells
NK cells are innate lymphocytes (CD16+, CD56+, no TCR) that provide rapid killing of virus-infected and tumor cells without prior sensitization. NK cell activity is regulated by the balance of activating and inhibitory receptors:
| Signal Type | Receptor | Ligand | Outcome |
|---|---|---|---|
| Inhibitory | KIR (killer immunoglobulin-like receptors) | MHC I on target cell | Inhibits killing (self-recognition) |
| Activating | NKG2D, natural cytotoxicity receptors | Stress ligands (MICA/MICB, ULBPs) | Triggers killing |
| Activating | FcγRIII (CD16) | IgG coating target cell | ADCC (antibody-dependent cellular cytotoxicity) |
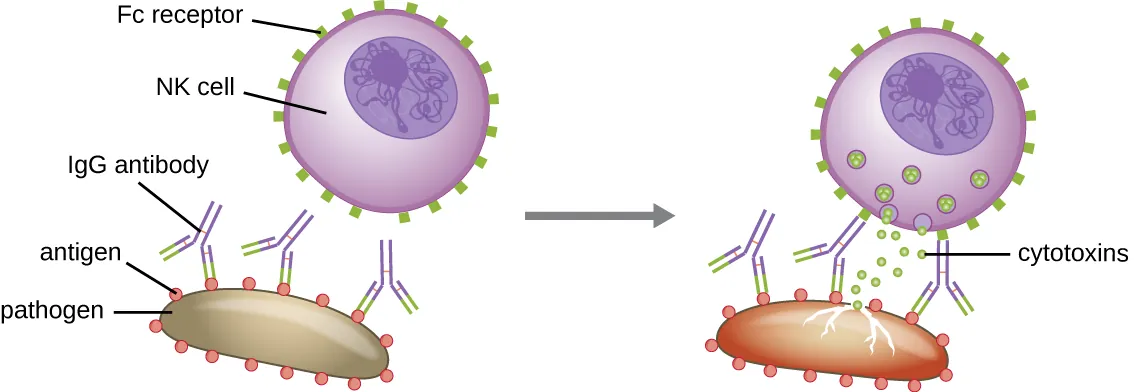
Missing-self hypothesis: NK cells kill cells that have lost MHC I expression (common in virally infected and tumor cells that downregulate MHC I to evade CTLs). Loss of the inhibitory KIR signal tips the balance toward activation → killing.
An autosomal recessive defect in the LYST gene causes impaired lysosomal trafficking, resulting in giant granules in neutrophils and NK cells. Patients present with recurrent pyogenic infections, partial oculocutaneous albinism, peripheral neuropathy, and defective NK cell function. Peripheral blood smear shows pathognomonic giant granules in neutrophils.
Perforin/Granzyme Killing in Detail
Both CTLs and NK cells use the perforin/granzyme pathway as their primary killing mechanism. The process is highly directional:
- Immune synapse formation: CTL/NK cell polarizes its microtubule-organizing center (MTOC) and lytic granules toward the target cell contact point
- Perforin release: Creates pores in target cell membrane (similar to MAC); also facilitates granzyme entry via endocytosis
- Granzyme B entry: Serine protease enters target cell and activates caspase cascade (caspase-3, -7) → apoptosis
- Serial killing: CTLs can detach and sequentially kill multiple target cells; this is why a relatively small number of CTLs can eliminate many virus-infected cells
Genetic defects in perforin (PRF1 mutations) or granule exocytosis machinery (Munc13-4, syntaxin-11) cause inability of CTLs and NK cells to kill target cells. This leads to unchecked immune activation, massive cytokine release (“cytokine storm”), and hemophagocytosis (macrophages engulfing blood cells). Presents with fever, hepatosplenomegaly, cytopenias, hyperferritinemia (>10,000), hypertriglyceridemia, and elevated soluble IL-2R. Treatment: etoposide-based chemotherapy followed by bone marrow transplant.
12 B-Cell Development & Activation
B cells develop in the bone marrow (hence "B") through a series of stages defined by immunoglobulin gene rearrangement status.
Stages of B-Cell Development
| Stage | Ig Status | Key Event |
|---|---|---|
| Pro-B cell | Heavy chain VDJ rearrangement begins | RAG-1/RAG-2 mediated; D-J first, then V-DJ |
| Pre-B cell | μ heavy chain + surrogate light chain (pre-BCR) | Successful heavy chain rearrangement; allelic exclusion ensures one heavy chain per cell |
| Immature B cell | Complete IgM on surface | Light chain rearrangement (κ first, then λ if needed); negative selection against self-antigens in bone marrow |
| Mature naïve B cell | IgM + IgD co-expressed on surface | Exits to periphery; circulates through secondary lymphoid organs awaiting antigen encounter |
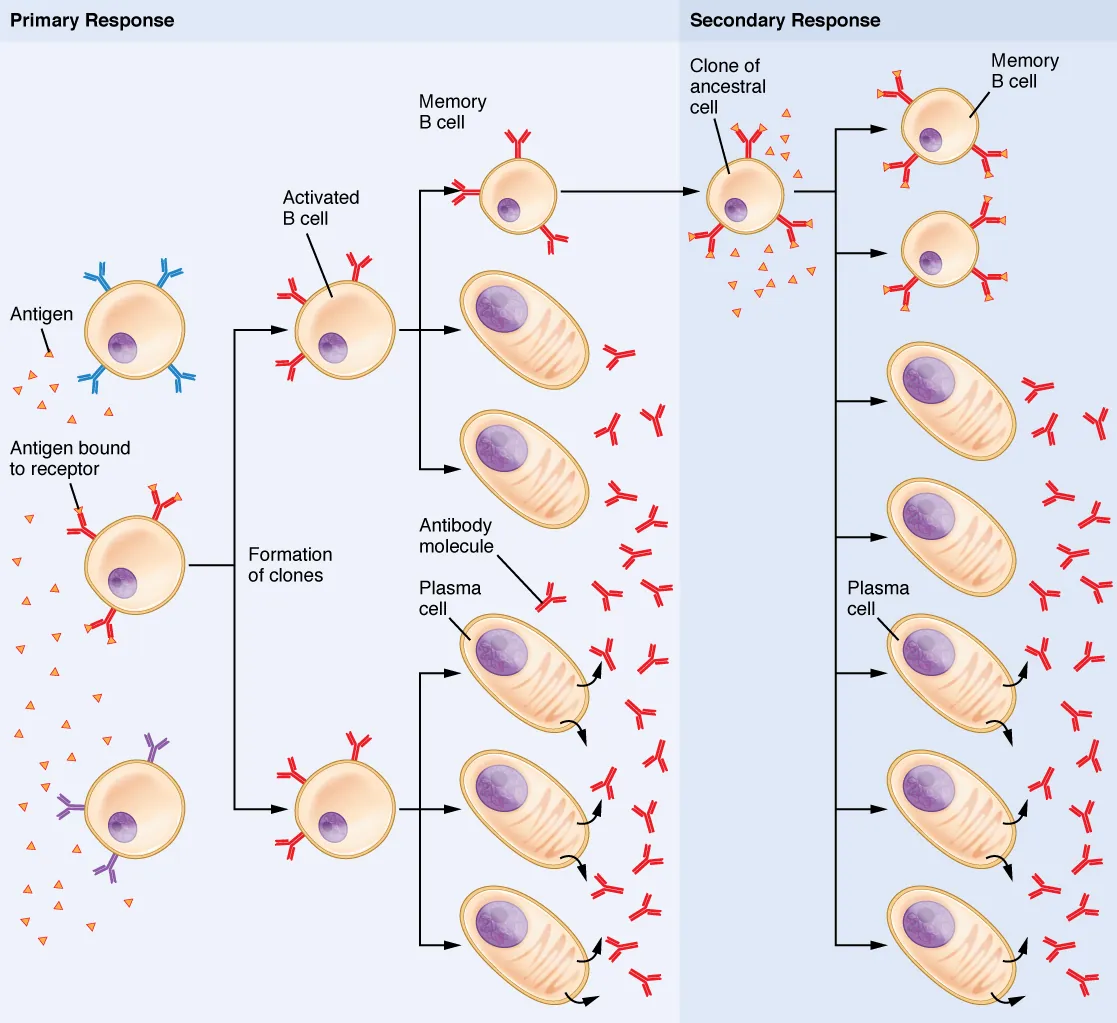
B-Cell Activation
B cells can be activated via two pathways:
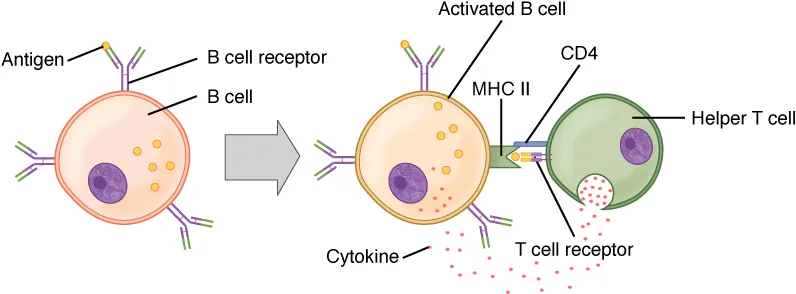
- T-cell dependent (TD): B cell internalizes antigen via BCR, processes it, and presents peptide on MHC II to a Tfh cell. Tfh provides co-stimulation (CD40L–CD40 interaction) and cytokines (IL-4, IL-21). This drives germinal center formation, affinity maturation, isotype switching, and memory cell generation.
- T-cell independent (TI): Polysaccharide antigens (e.g., bacterial capsules) cross-link multiple BCRs simultaneously, activating B cells without T-cell help. Produces mainly IgM (no isotype switching), no affinity maturation, weak memory. This is why polysaccharide vaccines (pneumococcal, meningococcal) are less effective in children <2 years old (immature marginal zone B cells).
13 Antibody Structure & Function
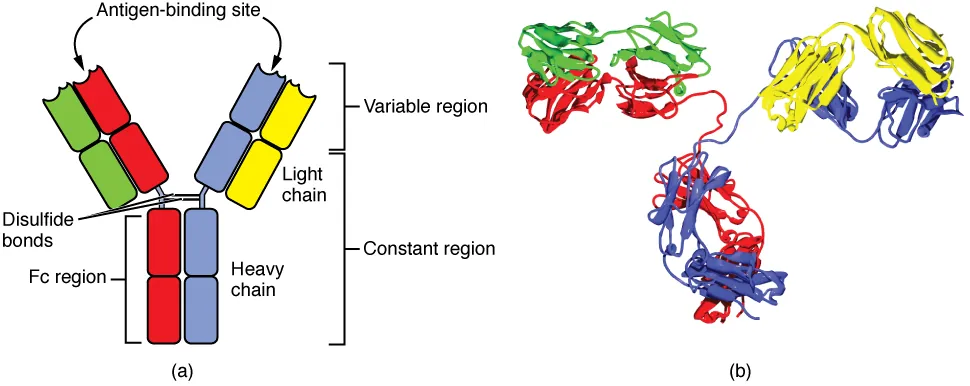
Basic Immunoglobulin Structure
Each antibody molecule consists of two identical heavy chains and two identical light chains (κ or λ), linked by disulfide bonds. Each chain has a variable (V) region (antigen binding) and constant (C) region (effector function). The Fab fragment (Fragment antigen-binding) contains the variable regions; the Fc fragment (Fragment crystallizable) contains the constant regions and mediates complement activation, opsonization, and placental transfer.
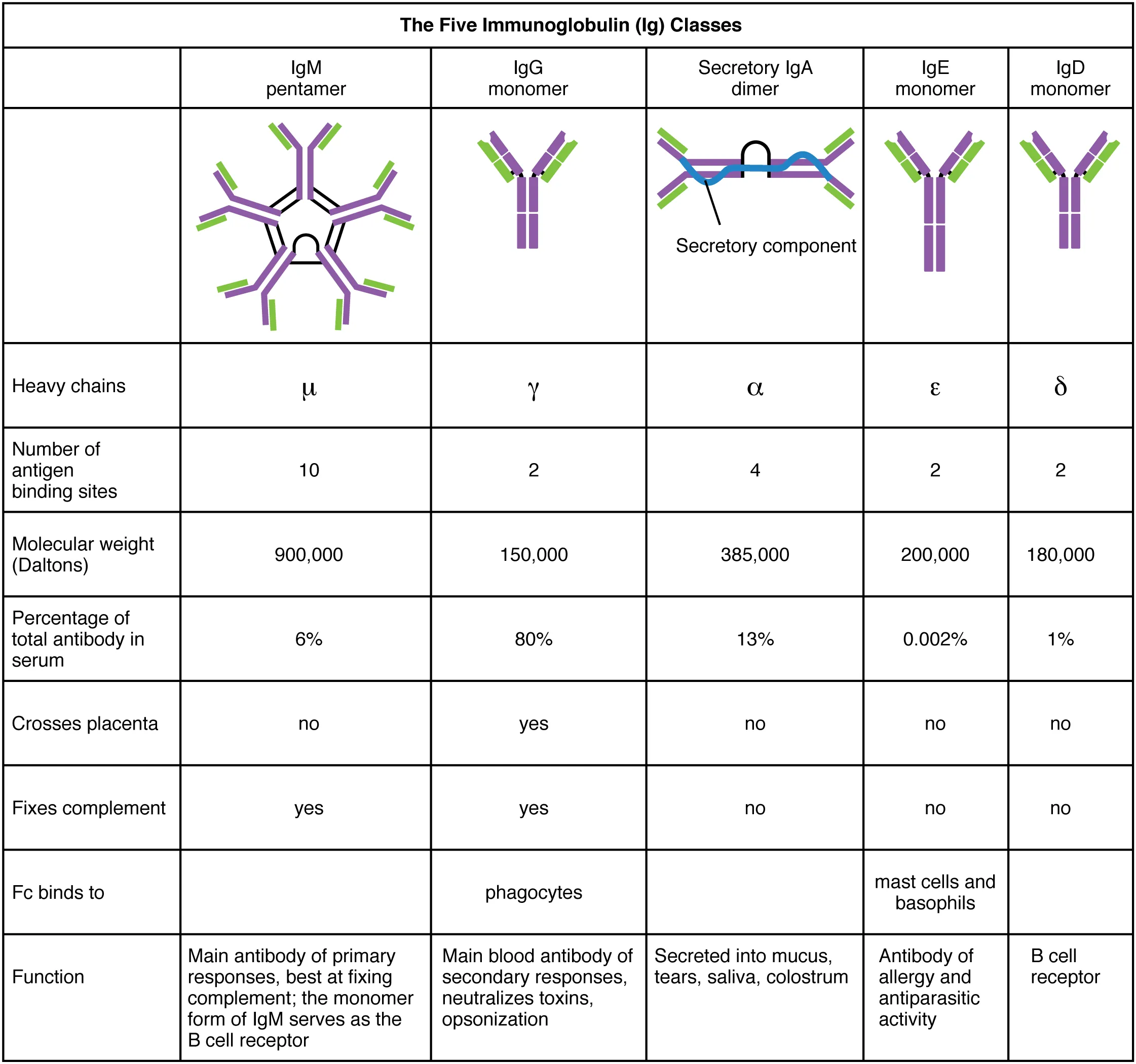
Immunoglobulin Isotypes
| Isotype | Heavy Chain | Structure | Serum % | Key Functions |
|---|---|---|---|---|
| IgG | γ | Monomer | 75–80% | Most abundant; crosses placenta (passive immunity to neonate); opsonization (FcγR); complement activation (classical); secondary immune response; ADCC |
| IgA | α | Dimer (secretory) or monomer (serum) | 10–15% | Mucosal immunity (#1 in secretions); secretory component protects from proteolysis; present in breast milk, saliva, tears, GI/respiratory secretions; does NOT activate complement |
| IgM | μ | Pentamer (serum) | 5–10% | First antibody produced in primary response; most efficient complement activator (one IgM pentamer can activate C1q); expressed on naïve B cells as monomer (BCR) |
| IgE | ε | Monomer | <0.01% | Binds FcεRI on mast cells/basophils; cross-linking triggers degranulation (Type I hypersensitivity); anti-helminth defense; elevated in atopic individuals and parasitic infections |
| IgD | δ | Monomer | <1% | Co-expressed with IgM on mature naïve B cells; role in B-cell activation; exact function not fully defined |
Antibody Diversity Mechanisms
| Mechanism | Description | When |
|---|---|---|
| VDJ recombination | Random selection of V, D, J gene segments (heavy chain) or V, J (light chain) | During B-cell development |
| Junctional diversity | Imprecise joining + N-nucleotide addition (by TdT) at VD and DJ junctions | During rearrangement |
| Combinatorial diversity | Random pairing of different heavy and light chains | During assembly |
| Somatic hypermutation | Point mutations in V regions during germinal center reaction; driven by AID (activation-induced cytidine deaminase) | After antigen exposure in germinal centers |
IgG Subclasses
| Subclass | % of Total IgG | Key Properties |
|---|---|---|
| IgG1 | 65% | Most abundant; activates complement; crosses placenta; opsonization |
| IgG2 | 25% | Responds to polysaccharide antigens (encapsulated bacteria); weak complement activation |
| IgG3 | 7% | Strongest complement activator of IgG subclasses; shortest half-life (~7 days vs. ~21 days for others) |
| IgG4 | 3% | Does not activate complement; associated with chronic antigen exposure; anti-inflammatory; IgG4-related disease causes fibroinflammatory lesions (autoimmune pancreatitis, retroperitoneal fibrosis) |
Fc Receptors & Effector Functions
| Receptor | Cells | Function |
|---|---|---|
| FcγRI (CD64) | Macrophages, neutrophils, dendritic cells | High-affinity IgG binding; opsonization and phagocytosis |
| FcγRIII (CD16) | NK cells, macrophages | Low-affinity IgG binding; mediates ADCC (antibody-dependent cellular cytotoxicity) |
| FcεRI | Mast cells, basophils | High-affinity IgE binding; cross-linking triggers degranulation (Type I hypersensitivity) |
| FcRn (neonatal Fc receptor) | Placental syncytiotrophoblast, endothelium | Transports IgG across placenta; recycles IgG (extends half-life to ~21 days) |
| Poly-Ig receptor | Mucosal epithelium | Transcytoses dimeric IgA (and pentameric IgM) across epithelium into lumen; cleaved to form secretory component |
14 Isotype Switching & Affinity Maturation
Isotype (Class) Switching
Isotype switching changes the antibody constant region (and thus the effector function) without altering antigen specificity. It requires:
- CD40L–CD40 interaction between Tfh cell and B cell (patients with CD40L deficiency → Hyper-IgM syndrome, X-linked; cannot switch from IgM)
- Cytokine signals that direct which isotype: IL-4 → IgE and IgG4; IFN-γ → IgG1/IgG3; TGF-β → IgA; IL-5 → IgA
- AID (activation-induced cytidine deaminase) — enzyme that initiates switch recombination at switch regions upstream of C genes. AID deficiency also causes hyper-IgM syndrome (autosomal recessive form)
Affinity Maturation
Occurs in germinal centers of secondary lymphoid organs. AID introduces somatic hypermutations in Ig variable regions. B cells with higher-affinity BCRs compete for limited antigen displayed on follicular dendritic cells (FDCs). Those with improved binding receive survival signals (selected); those with lower affinity undergo apoptosis. This iterative process progressively increases antibody affinity over the course of an immune response.
Dark zone: Rapidly proliferating centroblasts undergo somatic hypermutation (SHM). Light zone: Centrocytes are tested for antigen affinity on FDCs and receive T-cell help; those with high affinity are selected to become plasma cells or memory B cells. The germinal center reaction is the basis for affinity maturation, isotype switching, and long-lived immunologic memory.
Immunologic Memory
The germinal center reaction generates two key long-lived populations:
| Cell Type | Location | Function | Duration |
|---|---|---|---|
| Memory B cells | Circulating, marginal zone, mucosal tissues | Rapidly differentiate into plasma cells upon re-exposure; already isotype-switched and affinity-matured | Decades (some lifelong) |
| Long-lived plasma cells | Bone marrow niches | Continuously secrete antibody without re-stimulation; maintain baseline serum antibody levels | Years to decades |
| Memory T cells | Circulating (TCM) and tissue-resident (TRM) | Rapid effector response on re-encounter; lower activation threshold; secrete cytokines faster | Decades |
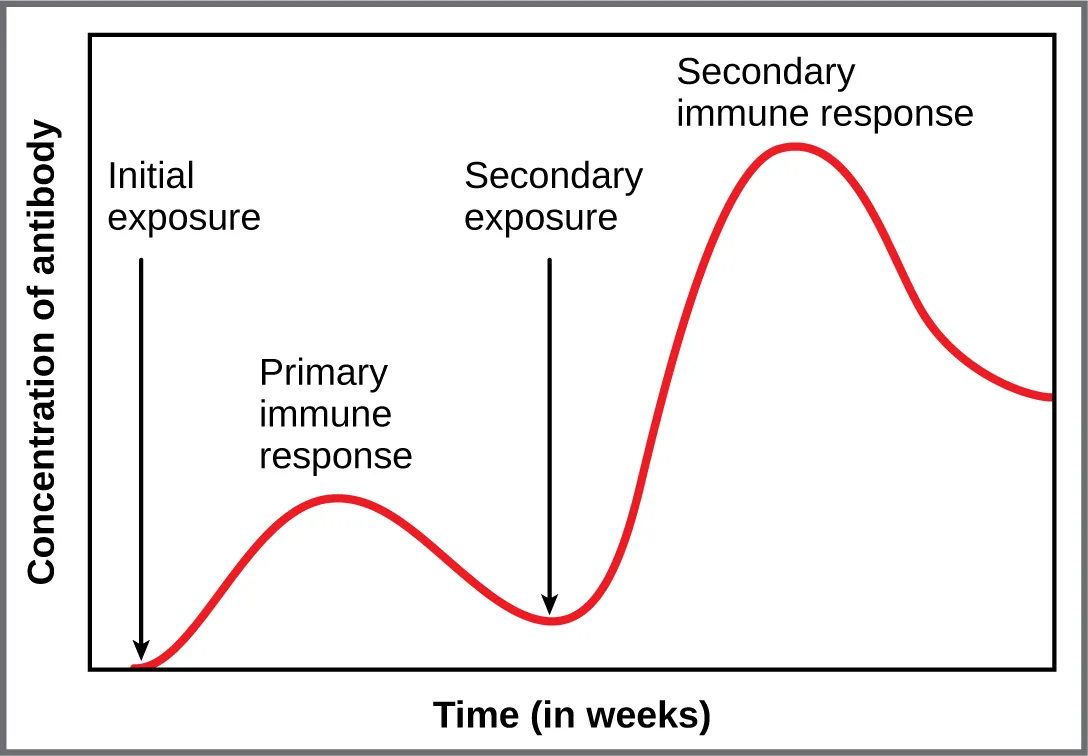
The secondary (anamnestic) immune response is faster (1–3 days vs. 7–10 days), stronger (higher antibody titer), predominantly IgG (class-switched), and higher affinity than the primary response. This is the immunologic basis for booster vaccinations.
15 Primary & Secondary Lymphoid Organs
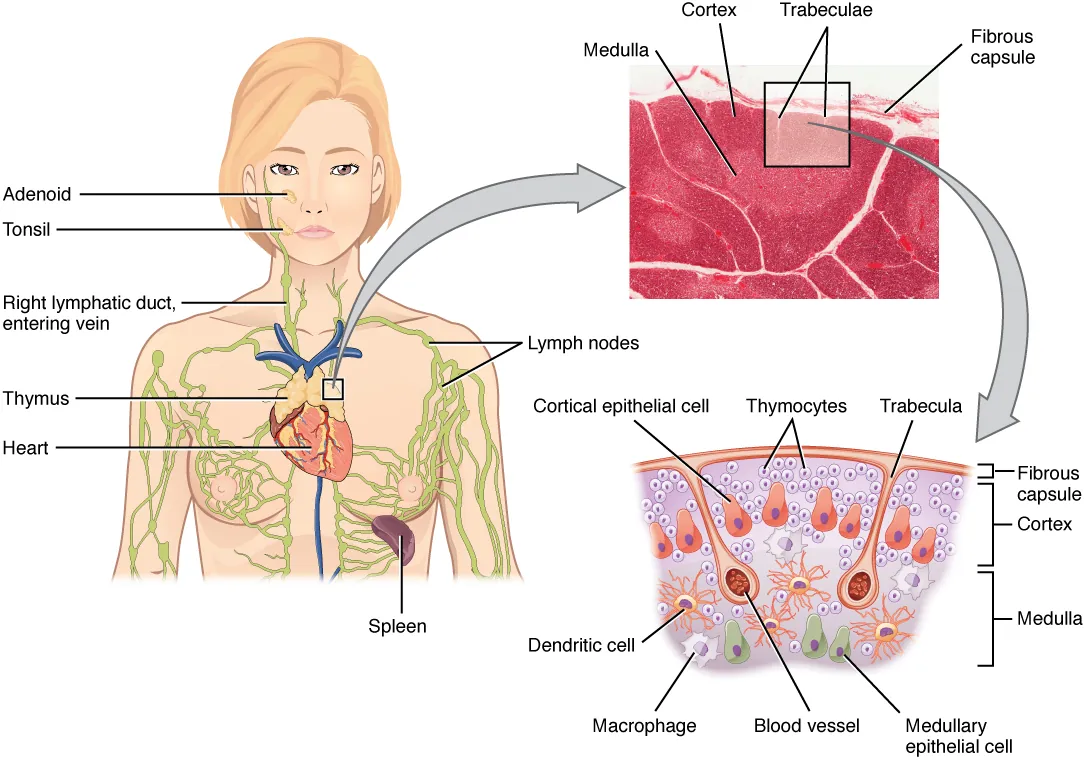
Primary (Central) Lymphoid Organs
| Organ | Function | Clinical Relevance |
|---|---|---|
| Bone marrow | Site of hematopoiesis; B-cell development and maturation; site of negative selection for B cells | Aplastic anemia, leukemia, myelodysplastic syndromes affect all blood cell lineages |
| Thymus | T-cell maturation, positive and negative selection; involutes after puberty (replaced by fat but retains some function) | DiGeorge syndrome (22q11.2 deletion) → thymic aplasia/hypoplasia → T-cell deficiency |
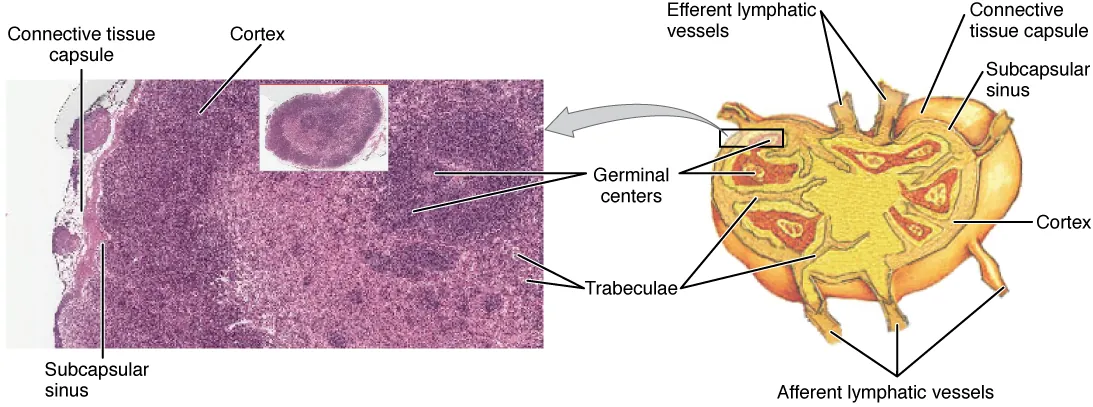
Secondary (Peripheral) Lymphoid Organs
| Organ | Structure & Function | Clinical Relevance |
|---|---|---|
| Lymph nodes | Cortex: B-cell follicles (primary = naïve; secondary = germinal centers). Paracortex: T-cell zone (contains high endothelial venules, HEVs). Medulla: medullary cords (plasma cells), medullary sinuses | Paracortex enlarges in T-cell responses (viral infections); follicular hyperplasia in B-cell responses. Paracortex absent in DiGeorge |
| Spleen | White pulp: PALS (T cells around central arteriole) + follicles/marginal zone (B cells). Red pulp: sinusoids, macrophages; filters blood, removes old/damaged RBCs and encapsulated organisms | Splenectomy → increased risk of infections with encapsulated organisms (SHiNE: S. pneumoniae, H. influenzae, N. meningitidis, E. coli, Salmonella); Howell-Jolly bodies on smear |
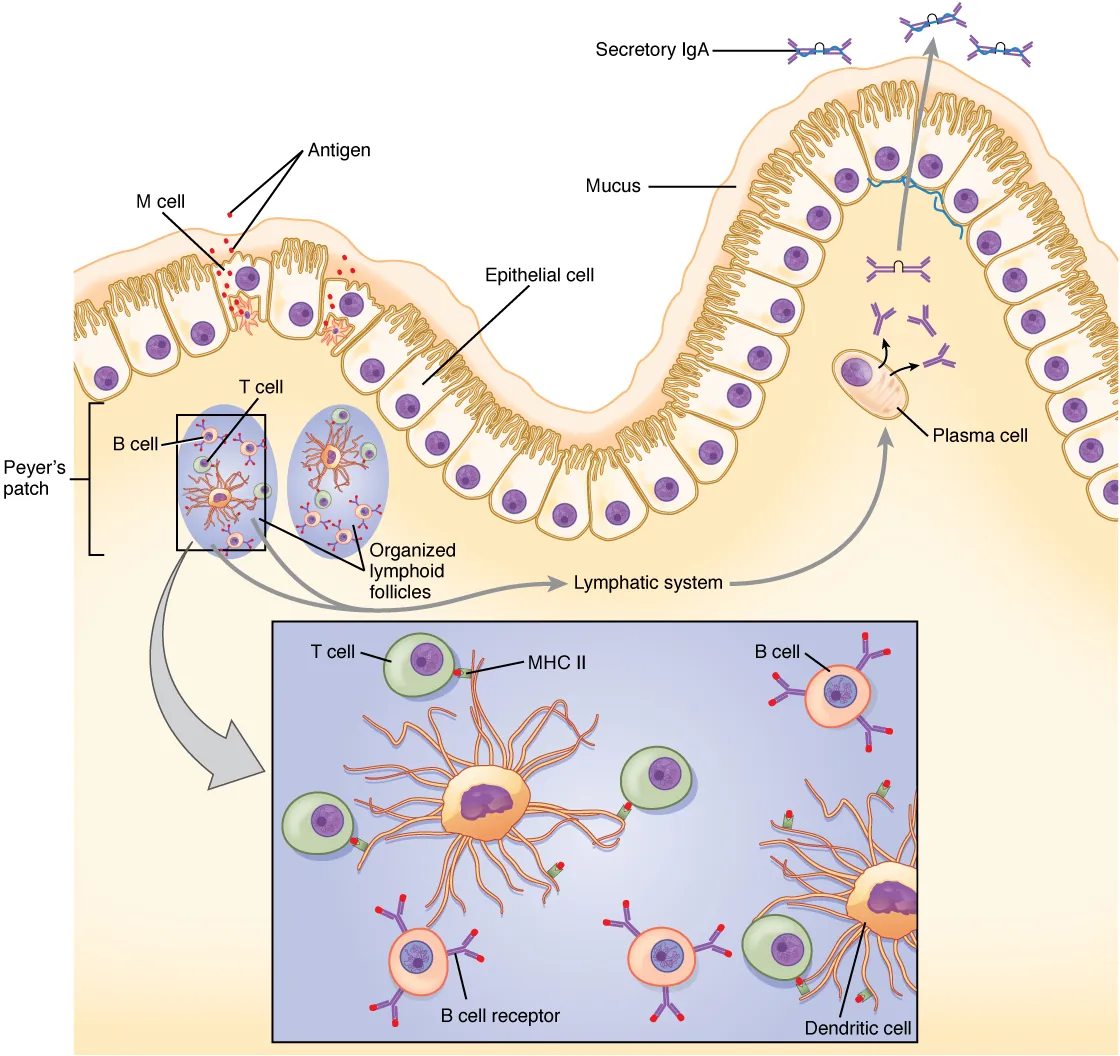
| MALT / GALT | Mucosal-associated lymphoid tissue; includes Peyer patches (ileum), tonsils, appendix, Waldeyer ring | IgA-secreting plasma cells in lamina propria; M cells sample luminal antigens for presentation to underlying lymphoid tissue |
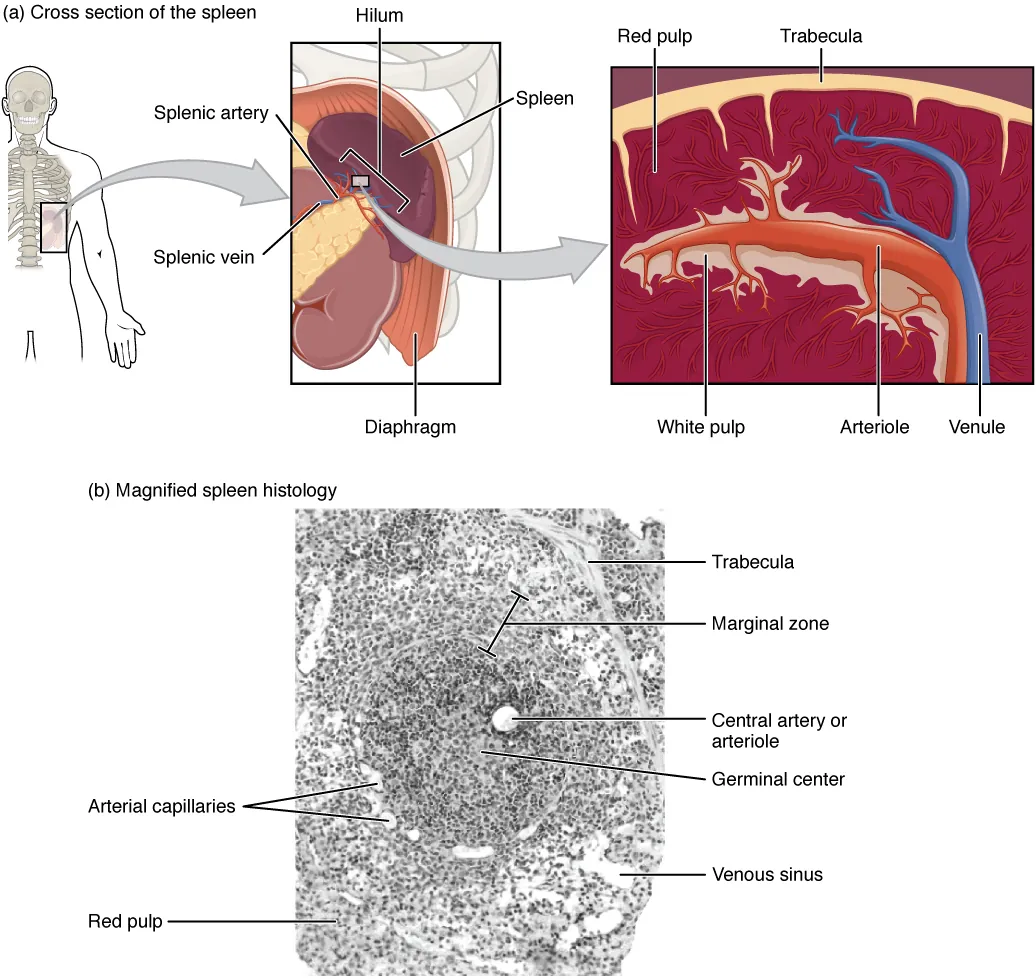
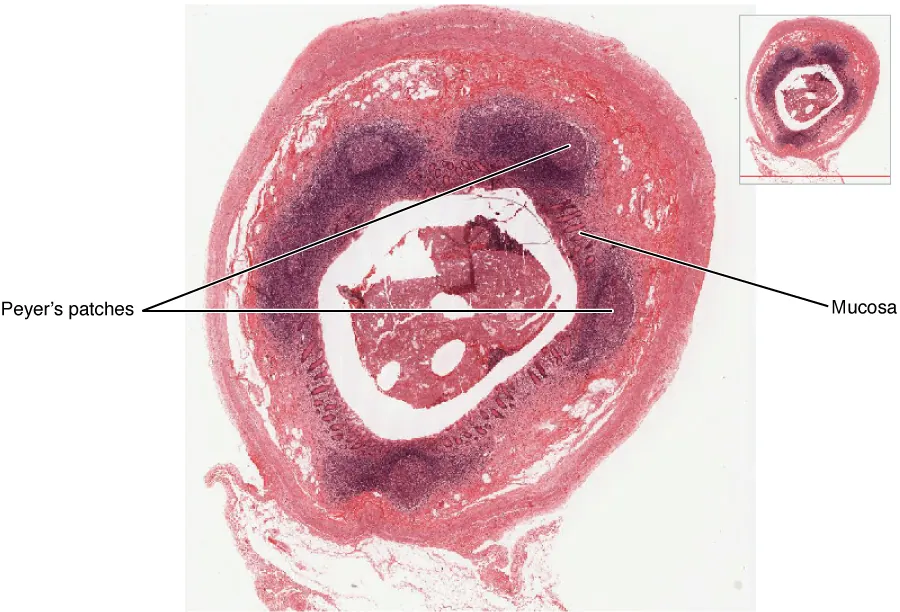
Lymphocyte Recirculation & Homing
Lymphocytes continuously recirculate between blood and lymphoid tissues. Naïve T cells enter lymph nodes via high endothelial venules (HEVs) using L-selectin (CD62L) and CCR7 (responds to CCL19/CCL21 chemokines produced by lymph node stroma). After activation, effector T cells downregulate L-selectin and CCR7 and upregulate tissue-specific homing receptors:
| Homing Receptor | Target Tissue | Ligand on Endothelium |
|---|---|---|
| CLA (cutaneous lymphocyte antigen) | Skin | E-selectin on dermal vessels |
| α4β7 integrin | Gut (Peyer patches, lamina propria) | MAdCAM-1 on gut endothelium |
| VLA-4 (α4β1) | CNS (across BBB), inflamed endothelium | VCAM-1 |
Natalizumab (anti-α4 integrin) blocks lymphocyte trafficking to CNS (used in MS) and gut (used in Crohn disease); risk of PML (JC virus) due to impaired CNS immune surveillance. Vedolizumab (anti-α4β7) selectively blocks gut homing (used in IBD) without CNS risk. Fingolimod (S1P receptor modulator) traps lymphocytes in lymph nodes (used in MS).
16 Immune Tolerance & Regulation
Central Tolerance
Occurs in primary lymphoid organs during lymphocyte development:
- T cells (thymus): Negative selection deletes strongly self-reactive T cells; some become Tregs (receptor editing is less common in T cells)
- B cells (bone marrow): Self-reactive immature B cells undergo receptor editing (new light chain rearrangement), clonal deletion (apoptosis), or clonal anergy (functional unresponsiveness)
Peripheral Tolerance
Mechanisms that silence self-reactive lymphocytes that escape central tolerance:
| Mechanism | Description |
|---|---|
| Anergy | T cell receives signal 1 (TCR engagement) without signal 2 (B7–CD28 co-stimulation) → becomes functionally unresponsive |
| Regulatory T cells (Tregs) | CD4+ CD25+ FoxP3+ cells suppress autoreactive T cells via IL-10, TGF-β, CTLA-4, and direct contact inhibition |
| Clonal deletion | Activation-induced cell death (AICD) via Fas/FasL pathway eliminates chronically stimulated T cells |
| Immune checkpoint molecules | CTLA-4 (competes with CD28 for B7, higher affinity; dampens T-cell activation); PD-1 (binds PD-L1/PD-L2 on target cells; induces T-cell exhaustion) |
| Immune privilege | Certain sites (brain, eye anterior chamber, testes, placenta) have limited immune access via blood-tissue barriers, local immunosuppressive factors (TGF-β, FasL expression) |
CTLA-4 acts early (during T-cell priming in lymph nodes); outcompetes CD28 for B7 binding and delivers inhibitory signals. PD-1 acts late (in peripheral tissues); binds PD-L1 on target/tumor cells and causes T-cell exhaustion. Blocking these checkpoints with antibodies (ipilimumab = anti-CTLA-4; pembrolizumab/nivolumab = anti-PD-1) unleashes anti-tumor T-cell responses but can cause immune-related adverse events (autoimmune colitis, hepatitis, thyroiditis).
Superantigens
Superantigens are microbial proteins that bypass normal antigen processing and bind directly to the outside of MHC II and the Vβ region of the TCR, non-specifically activating a large fraction (2–20%) of all T cells simultaneously. This triggers massive cytokine release (“cytokine storm”) — TNF-α, IL-1, IL-2, IFN-γ — leading to high fever, hypotension, rash, and potentially multi-organ failure.
| Superantigen | Source | Disease |
|---|---|---|
| TSST-1 (Toxic Shock Syndrome Toxin-1) | S. aureus | Toxic shock syndrome (tampon-associated or wound-associated) |
| Exotoxin A (SpeA) | S. pyogenes (Group A Strep) | Streptococcal toxic shock syndrome, scarlet fever |
| Staphylococcal enterotoxins (SE-A through SE-E) | S. aureus | Staphylococcal food poisoning (rapid-onset vomiting and diarrhea) |
Normal antigens are processed into peptides and presented within the MHC groove to the TCR CDR3 region — activating only ~0.001% of T cells (clonal response). Superantigens bind outside the groove to the Vβ region of TCR and the lateral face of MHC II, activating all T cells sharing that Vβ type (2–20% of all T cells). This explains the massive, polyclonal T-cell activation and cytokine storm in superantigen-mediated diseases.
17 Cytokines & Signaling Networks
Major Cytokines & Their Functions
| Cytokine | Main Source | Key Functions |
|---|---|---|
| IL-1 | Macrophages | Fever, acute-phase proteins, endothelial activation; co-stimulates T cells |
| IL-2 | Th1 cells | T-cell proliferation and survival; promotes Treg development; used therapeutically (high dose in melanoma/RCC) |
| IL-3 | T cells | Multi-lineage hematopoietic growth factor (supports growth of all bone marrow progenitors) |
| IL-4 | Th2 cells, mast cells | B-cell class switch to IgE and IgG4; Th2 differentiation; inhibits Th1 |
| IL-5 | Th2 cells | Eosinophil production, activation, and survival; IgA class switching |
| IL-6 | Macrophages, Th2 | Fever, acute-phase proteins (CRP), B-cell differentiation to plasma cells; Th17 differentiation |
| IL-8 (CXCL8) | Macrophages | Neutrophil chemotaxis (most important neutrophil chemoattractant among cytokines) |
| IL-10 | Tregs, Th2, macrophages | Anti-inflammatory; suppresses Th1 and macrophage function; inhibits APC activity |
| IL-12 | Dendritic cells, macrophages | Th1 differentiation; NK cell activation; induces IFN-γ production |
| IL-17 | Th17 cells | Neutrophil recruitment; pro-inflammatory; mucosal defense; implicated in autoimmunity |
| IL-23 | Dendritic cells, macrophages | Th17 maintenance and expansion; target in psoriasis treatment (ustekinumab targets shared p40 subunit of IL-12/23) |
| IFN-α/β | Virus-infected cells (all nucleated cells) | Antiviral state in neighboring cells; upregulate MHC I; activate NK cells; Type I interferons |
| IFN-γ | Th1, NK cells, CD8+ T cells | Macrophage activation (most potent); upregulates MHC I & II; promotes Th1; antiviral; Type II interferon |
| TNF-α | Macrophages, T cells | Sepsis, cachexia, endothelial activation, leukocyte recruitment; major mediator of Gram-negative shock |
| TGF-β | Many cell types | Anti-inflammatory; promotes Treg and Th17 differentiation (context-dependent); class switch to IgA; wound healing |
Anti-TNF: infliximab, adalimumab, etanercept (RA, IBD, psoriasis; risk of TB reactivation). Anti-IL-1: anakinra (gout, CAPS, Still disease). Anti-IL-6R: tocilizumab (RA, giant cell arteritis, cytokine release syndrome). Anti-IL-5: mepolizumab (eosinophilic asthma). Anti-IL-4R: dupilumab (atopic dermatitis, asthma). Anti-IL-17A: secukinumab (psoriasis, ankylosing spondylitis).
Cytokine Signaling Pathways
| Pathway | Cytokines | Mechanism | Clinical Significance |
|---|---|---|---|
| JAK-STAT | Most interleukins, IFNs, growth factors | Cytokine receptor activates JAK (Janus kinase) → phosphorylates STAT → STAT dimerizes, translocates to nucleus, activates gene transcription | JAK inhibitors (tofacitinib, baricitinib, ruxolitinib) used in RA, myelofibrosis, GvHD, atopic dermatitis |
| NF-κB | TNF-α, IL-1, TLR ligands | Activates transcription of pro-inflammatory genes (cytokines, adhesion molecules, MHC) | Constitutive NF-κB activation in many cancers and autoimmune diseases; corticosteroids inhibit NF-κB as part of their anti-inflammatory mechanism |
| MAPK/ERK | Growth factors (EGF, PDGF), cytokines | RAS → RAF → MEK → ERK cascade; promotes cell proliferation and differentiation | Oncogenic RAS mutations in many cancers; MEK inhibitors (trametinib) used in melanoma |
| PI3K/AKT/mTOR | IL-2, growth factors | Promotes cell survival, proliferation, metabolism | Sirolimus inhibits mTOR (immunosuppression); PI3K inhibitors (idelalisib) in CLL |
Chemokines & Their Receptors
| Chemokine | Receptor | Function |
|---|---|---|
| IL-8 (CXCL8) | CXCR1, CXCR2 | Neutrophil chemotaxis (acute inflammation) |
| CCL2 (MCP-1) | CCR2 | Monocyte recruitment to inflammation sites |
| CCL3, CCL4, CCL5 (RANTES) | CCR5 | T cell, monocyte, NK cell recruitment; CCR5 is HIV co-receptor |
| CXCL12 (SDF-1) | CXCR4 | Hematopoietic stem cell homing to bone marrow; CXCR4 is HIV co-receptor (late) |
| CCL19, CCL21 | CCR7 | Naïve T cell and dendritic cell homing to lymph nodes (via HEVs) |
| CCL11 (eotaxin) | CCR3 | Eosinophil recruitment (allergic inflammation, parasitic infection) |
18 Hypersensitivity Type I — Immediate (IgE-Mediated)
Type I hypersensitivity is an immediate reaction (minutes) mediated by IgE antibodies bound to mast cells and basophils via FcεRI. On re-exposure, antigen cross-links surface IgE, triggering degranulation and release of preformed mediators (histamine, tryptase, heparin) and newly synthesized mediators (leukotrienes, prostaglandins, PAF, cytokines).
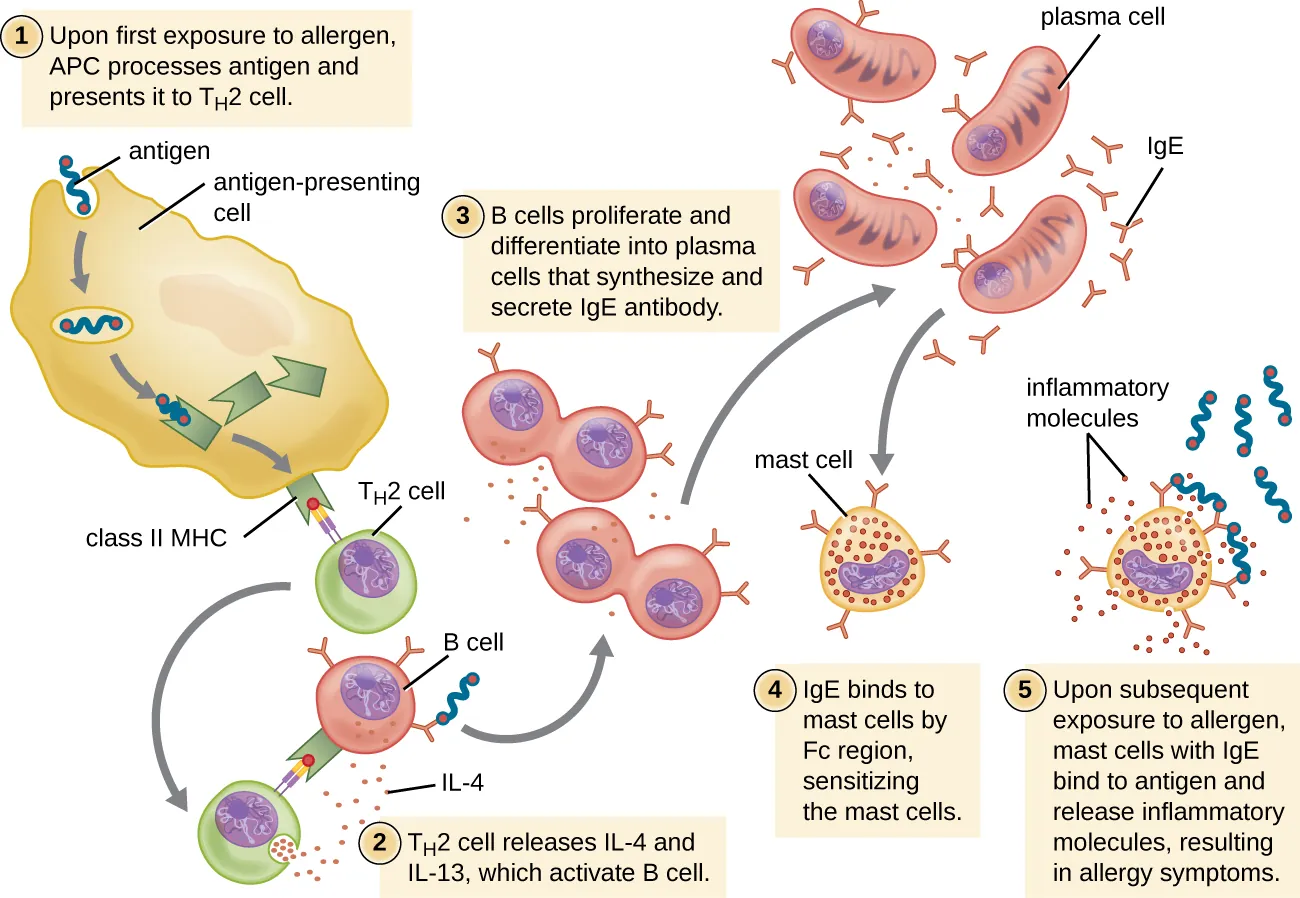
Phases of Type I Reaction
| Phase | Timing | Mediators | Effects |
|---|---|---|---|
| Sensitization | First exposure (days–weeks) | IL-4 drives IgE class switch; IgE binds FcεRI on mast cells | No symptoms; mast cells are now “armed” |
| Early (immediate) | Minutes after re-exposure | Histamine, tryptase, heparin (preformed granule contents) | Vasodilation, vascular permeability, bronchospasm, mucus secretion |
| Late | 4–8 hours | Leukotrienes (LTC4/D4/E4), prostaglandins, PAF, cytokines (IL-4, IL-5, TNF-α) | Eosinophil recruitment, persistent inflammation, tissue damage |
Clinical Examples
Anaphylaxis (peanuts, bee stings, penicillin, latex), allergic asthma, allergic rhinitis (hay fever), urticaria (hives), atopic dermatitis (eczema), food allergies.
Mast Cell Mediators
| Mediator | Type | Actions |
|---|---|---|
| Histamine | Preformed (granule) | Vasodilation (H1, H2), increased vascular permeability (H1), bronchospasm (H1), gastric acid secretion (H2), pruritus |
| Tryptase | Preformed (granule) | Serine protease; serum marker of mast cell degranulation (diagnostic for anaphylaxis) |
| Heparin | Preformed (granule) | Anticoagulant; binds to granule matrix proteoglycans |
| LTC4, LTD4, LTE4 | Newly synthesized (lipoxygenase) | Bronchoconstriction (1,000× more potent than histamine), vasoconstriction, mucus secretion; target of montelukast and zileuton |
| PGD2 | Newly synthesized (COX) | Vasodilation, bronchospasm, neutrophil/Th2 chemotaxis |
| PAF (platelet-activating factor) | Newly synthesized | Platelet aggregation, bronchospasm, hypotension; potent mediator in anaphylactic shock |
| TNF-α, IL-4, IL-5, IL-13 | Newly synthesized (cytokines) | Late-phase inflammation, eosinophil recruitment, IgE class switching |
Treatment
Anaphylaxis: Epinephrine IM (first-line, lifesaving); antihistamines (H1 and H2 blockers); corticosteroids (prevent late phase); IV fluids for hypotension. Chronic allergic disease: Antihistamines, inhaled corticosteroids, leukotriene receptor antagonists (montelukast), cromolyn (mast cell stabilizer), omalizumab (anti-IgE monoclonal antibody), allergen immunotherapy (desensitization).
19 Hypersensitivity Type II — Antibody-Mediated Cytotoxicity
Type II hypersensitivity involves IgG or IgM antibodies directed against antigens on cell surfaces or extracellular matrix. Damage occurs via complement activation (MAC lysis), opsonization and phagocytosis, or antibody-dependent cellular cytotoxicity (ADCC by NK cells).
Mechanisms & Examples
| Subtype | Mechanism | Examples |
|---|---|---|
| Cytotoxic (cell destruction) | Antibody binds cell surface antigen → complement activation (MAC) and/or opsonization → cell lysis/phagocytosis | Autoimmune hemolytic anemia, transfusion reactions (ABO incompatibility), hemolytic disease of the newborn (Rh), ITP (anti-platelet antibodies), Goodpasture syndrome (anti-GBM) |
| Anti-receptor (stimulatory) | Antibody binds and activates receptor | Graves disease (anti-TSH receptor → thyroid stimulation and hyperthyroidism) |
| Anti-receptor (inhibitory) | Antibody binds and blocks receptor | Myasthenia gravis (anti-AChR → NMJ blockade); pernicious anemia (anti-intrinsic factor) |
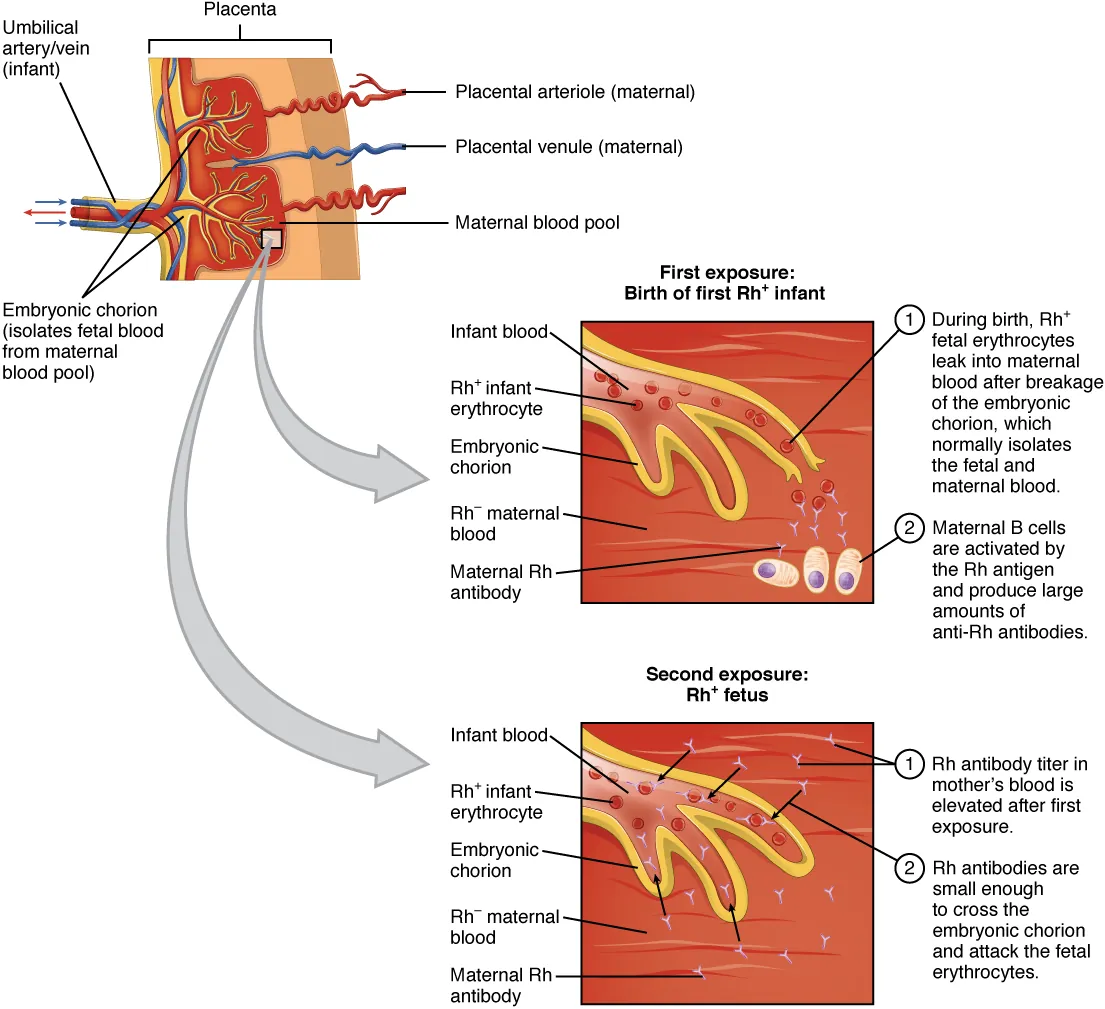
Type II Hypersensitivity in Blood Transfusion
| Reaction | Mechanism | Timing | Features |
|---|---|---|---|
| ABO incompatibility (acute hemolytic) | Pre-existing IgM anti-A or anti-B → complement activation → intravascular hemolysis | Minutes | Fever, flank pain, hemoglobinuria, DIC, renal failure; most dangerous transfusion reaction |
| Rh incompatibility (delayed hemolytic) | IgG anti-D (from prior sensitization) → extravascular hemolysis in spleen | Days–weeks | Gradual Hgb drop, indirect hyperbilirubinemia, positive DAT |
| Hemolytic disease of newborn (erythroblastosis fetalis) | Maternal IgG anti-D crosses placenta → attacks fetal Rh+ RBCs | In utero / neonatal | Hydrops fetalis, jaundice, kernicterus; prevented by RhoGAM (anti-D Ig) at 28 weeks and within 72 hours of delivery |
Anti-GBM antibodies (IgG against type IV collagen α3 chain) cause rapidly progressive glomerulonephritis and pulmonary hemorrhage. Immunofluorescence shows linear IgG deposition along the glomerular basement membrane (vs. granular “lumpy-bumpy” in Type III). Treatment: plasmapheresis (remove antibodies) + cyclophosphamide + corticosteroids.
20 Hypersensitivity Type III — Immune Complex
Type III hypersensitivity results from deposition of antigen–antibody (immune) complexes in tissues, particularly blood vessel walls, joints, and kidneys. Deposited complexes activate complement, generating C3a and C5a (recruit neutrophils) → neutrophil-mediated tissue damage via lysosomal enzyme release and reactive oxygen species.
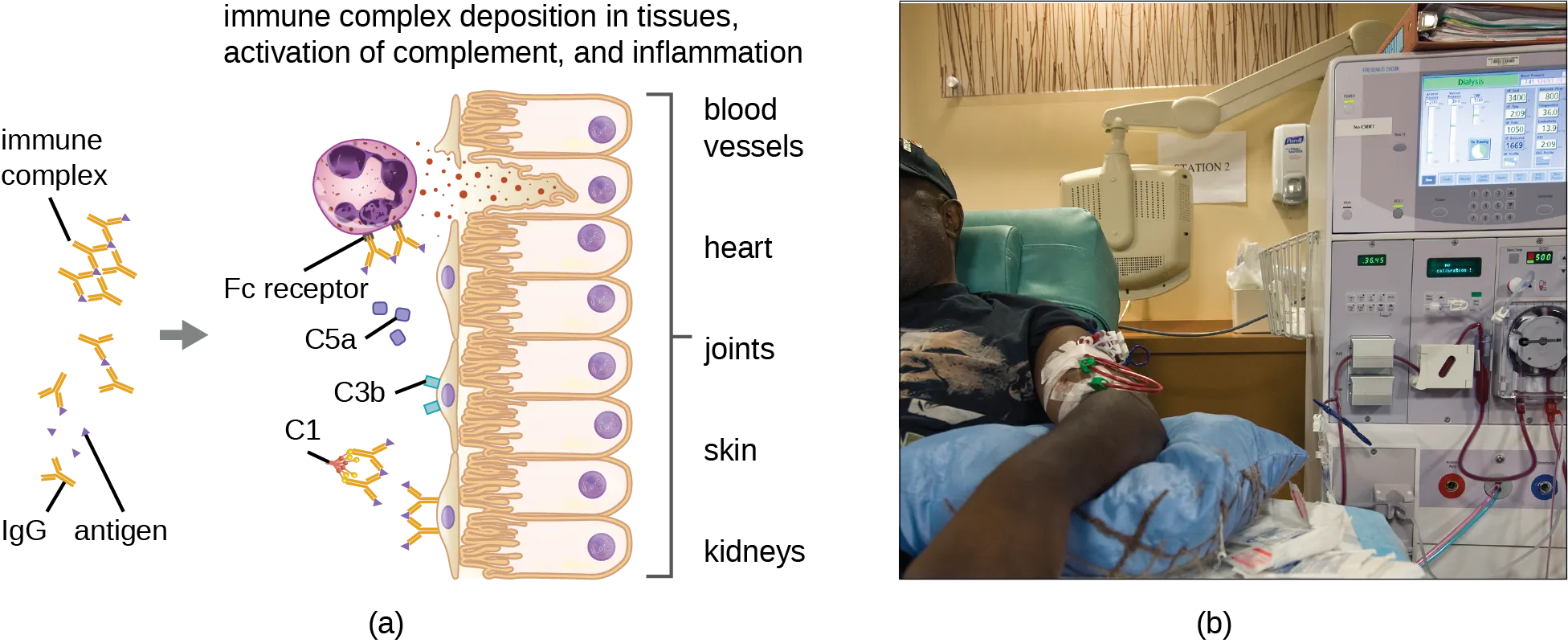
Key Clinical Examples
| Disease | Antigen Source | Clinical Features |
|---|---|---|
| Serum sickness | Foreign serum proteins (antithymocyte globulin, monoclonal antibodies) | Fever, urticaria, arthralgias, lymphadenopathy, proteinuria; 7–14 days after exposure |
| Arthus reaction | Local antigen injection in previously sensitized individual | Localized vasculitis, edema, necrosis at injection site; 4–10 hours |
| Polyarteritis nodosa | Hepatitis B surface antigen | Necrotizing vasculitis of medium vessels; renal, GI, peripheral nerve involvement |
| SLE nephritis | Anti-dsDNA immune complexes | Glomerulonephritis with granular (“lumpy-bumpy”) IF; wire-loop lesion on biopsy |
| Post-streptococcal GN | Streptococcal antigens | Nephritic syndrome 2–3 weeks after GAS pharyngitis; subepithelial “humps” on EM; granular IF (IgG + C3) |
| Hypersensitivity pneumonitis | Inhaled organic antigens (mold, bird droppings) | Farmer’s lung, bird fancier’s lung; mixed Type III and IV reaction |
Pathogenesis of Immune Complex Disease
Immune complex deposition depends on the size and solubility of the complexes. Small complexes remain soluble and are cleared. Very large complexes are quickly phagocytosed. Intermediate-sized complexes are the most pathogenic — they deposit in vessel walls, glomeruli, and joints, activate complement (C3a, C5a), recruit neutrophils, and cause tissue damage via lysosomal enzyme release and reactive oxygen species. The classic triad of immune complex disease is vasculitis, glomerulonephritis, and arthritis.
21 Hypersensitivity Type IV — Delayed-Type
Type IV hypersensitivity is the only type that is T-cell mediated (not antibody-mediated). It is “delayed” because it takes 24–72 hours to develop, requiring T-cell sensitization, migration, and cytokine-mediated tissue damage.
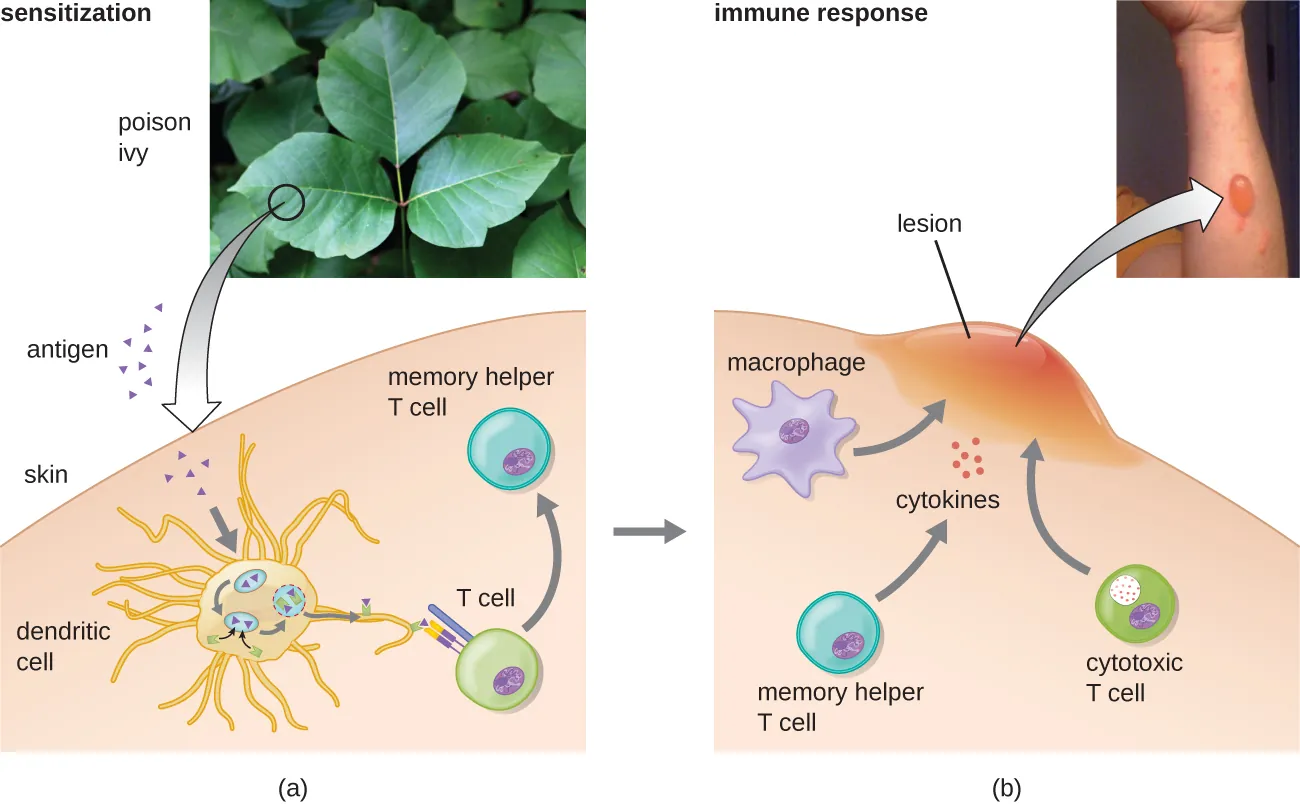
Subtypes of Type IV
| Subtype | Effector Cells | Mechanism | Examples |
|---|---|---|---|
| IVa (contact/DTH) | CD4+ Th1 cells, macrophages | Th1 cells secrete IFN-γ → macrophage activation → granulomatous inflammation | Tuberculin (PPD) skin test, contact dermatitis (poison ivy, nickel), granulomas (TB, sarcoidosis, Crohn) |
| IVb (T-cell mediated cytotoxicity) | CD8+ cytotoxic T cells | Direct killing of target cells expressing foreign or altered self-antigen on MHC I | Graft rejection (acute cellular), Stevens-Johnson syndrome/TEN (drug-induced), type 1 diabetes (islet cell destruction) |
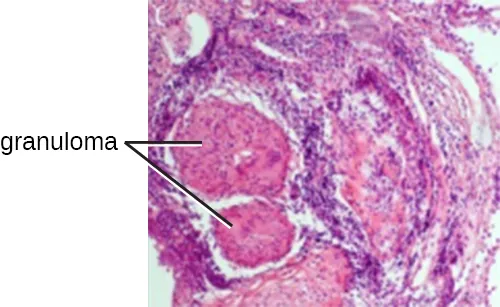
Granulomatous Inflammation
Chronic Type IV response to persistent antigens leads to granuloma formation: epithelioid macrophages (activated macrophages with abundant pink cytoplasm) surrounded by a collar of lymphocytes, often with multinucleated giant cells (Langhans type = peripheral nuclei horseshoe arrangement; foreign body type = haphazard nuclei).
| Disease | Granuloma Type | Key Feature |
|---|---|---|
| Tuberculosis | Caseating (central necrosis) | Acid-fast bacilli; Ghon complex (primary); cavitary lesions (reactivation) |
| Sarcoidosis | Non-caseating | Bilateral hilar lymphadenopathy; elevated ACE; hypercalcemia (1α-hydroxylase in macrophages) |
| Crohn disease | Non-caseating | Transmural inflammation; skip lesions; cobblestone mucosa |
| Fungal infections | Caseating or non-caseating | Histoplasma, Coccidioides, Blastomyces |
| Foreign body reaction | Foreign body type giant cells | Suture material, talc, silicone, beryllium |
| Cat-scratch disease | Suppurative (stellate microabscesses) | Bartonella henselae; regional lymphadenopathy |
Intradermal injection of purified protein derivative (PPD). A positive result (induration ≥5–15 mm at 48–72 hours, depending on risk group) indicates prior sensitization to mycobacterial antigens (prior TB infection, BCG vaccination, or atypical mycobacteria). The reaction is mediated by Th1 cells and macrophages — the classic example of Type IV hypersensitivity. Anergic patients (HIV, immunosuppression) may have false-negative PPD despite active TB.
Additional Type IV Examples in Clinical Medicine
| Disease / Reaction | Antigen | Mechanism |
|---|---|---|
| Contact dermatitis (poison ivy) | Urushiol (hapten) bound to skin proteins | Th1-mediated; epidermal Langerhans cells present antigen; rash at 24–72 hours |
| Contact dermatitis (nickel) | Nickel ions bind skin proteins | Th1-mediated; common cause of ear piercing dermatitis |
| Tuberculin (PPD) test | Mycobacterial proteins | Th1/macrophage-mediated induration at 48–72 hours |
| Hashimoto thyroiditis | Thyroid antigens (thyroglobulin, TPO) | CD8+ T cell destruction of thyroid follicular cells; anti-TPO and anti-thyroglobulin antibodies |
| Type 1 diabetes mellitus | Pancreatic β-cell antigens (GAD65, insulin, IA-2) | CD8+ T cell destruction of β-cells (insulitis); HLA-DR3/DR4 associated |
| Multiple sclerosis | Myelin antigens (MBP, MOG, PLP) | Th1/Th17-mediated demyelination in CNS; HLA-DR2 associated |
| Graft rejection (acute cellular) | Donor MHC molecules | Recipient CD4+ and CD8+ T cells attack donor tissue |
| Stevens-Johnson syndrome / TEN | Drug-modified self-proteins | CD8+ cytotoxic T cells and NK cells kill keratinocytes via Fas/FasL and granulysin |
22 Autoimmune Diseases & Autoantibodies
Autoimmune diseases arise from a loss of self-tolerance. They are classified as organ-specific (targeting a single organ) or systemic (affecting multiple organs).
High-Yield Autoantibody Associations
| Autoantibody | Disease |
|---|---|
| Anti-dsDNA | SLE (specific; correlates with disease activity and nephritis) |
| Anti-Smith (anti-Sm) | SLE (most specific, but less sensitive) |
| ANA (antinuclear antibody) | SLE (sensitive ~95%, but not specific; also positive in RA, scleroderma, Sjögren) |
| Anti-histone | Drug-induced lupus (hydralazine, isoniazid, procainamide, phenytoin, minocycline) |
| Anti-CCP (anti-cyclic citrullinated peptide) | Rheumatoid arthritis (most specific) |
| Rheumatoid factor (RF) | Rheumatoid arthritis (IgM against Fc of IgG; sensitive but not specific) |
| Anti-centromere | Limited scleroderma (CREST syndrome) |
| Anti-Scl-70 (anti-topoisomerase I) | Diffuse scleroderma (systemic sclerosis) |
| Anti-SSA (Ro) / Anti-SSB (La) | Sjögren syndrome; neonatal lupus (anti-Ro crosses placenta → congenital heart block) |
| Anti-Jo-1 (anti-histidyl tRNA synthetase) | Polymyositis/dermatomyositis with ILD (antisynthetase syndrome) |
| Anti-U1 RNP | Mixed connective tissue disease (MCTD) |
| c-ANCA (anti-PR3) | Granulomatosis with polyangiitis (GPA, formerly Wegener) |
| p-ANCA (anti-MPO) | Microscopic polyangiitis, eosinophilic GPA (Churg–Strauss) |
| Anti-GBM | Goodpasture syndrome |
| Anti-phospholipid (anticardiolipin, anti-β2GPI, lupus anticoagulant) | Antiphospholipid syndrome (recurrent thrombosis, pregnancy loss) |
| Anti-TSH receptor | Graves disease (stimulating antibody) |
| Anti-TPO, anti-thyroglobulin | Hashimoto thyroiditis |
| Anti-AChR | Myasthenia gravis (~85%) |
| Anti-MuSK | Myasthenia gravis (AChR-negative subset) |
| Anti-VGCC | Lambert–Eaton myasthenic syndrome |
| Anti-endomysial / Anti-tTG (tissue transglutaminase) | Celiac disease |
| Anti-mitochondrial (AMA) | Primary biliary cholangitis (PBC) |
| Anti-smooth muscle (ASMA) | Autoimmune hepatitis (Type 1) |
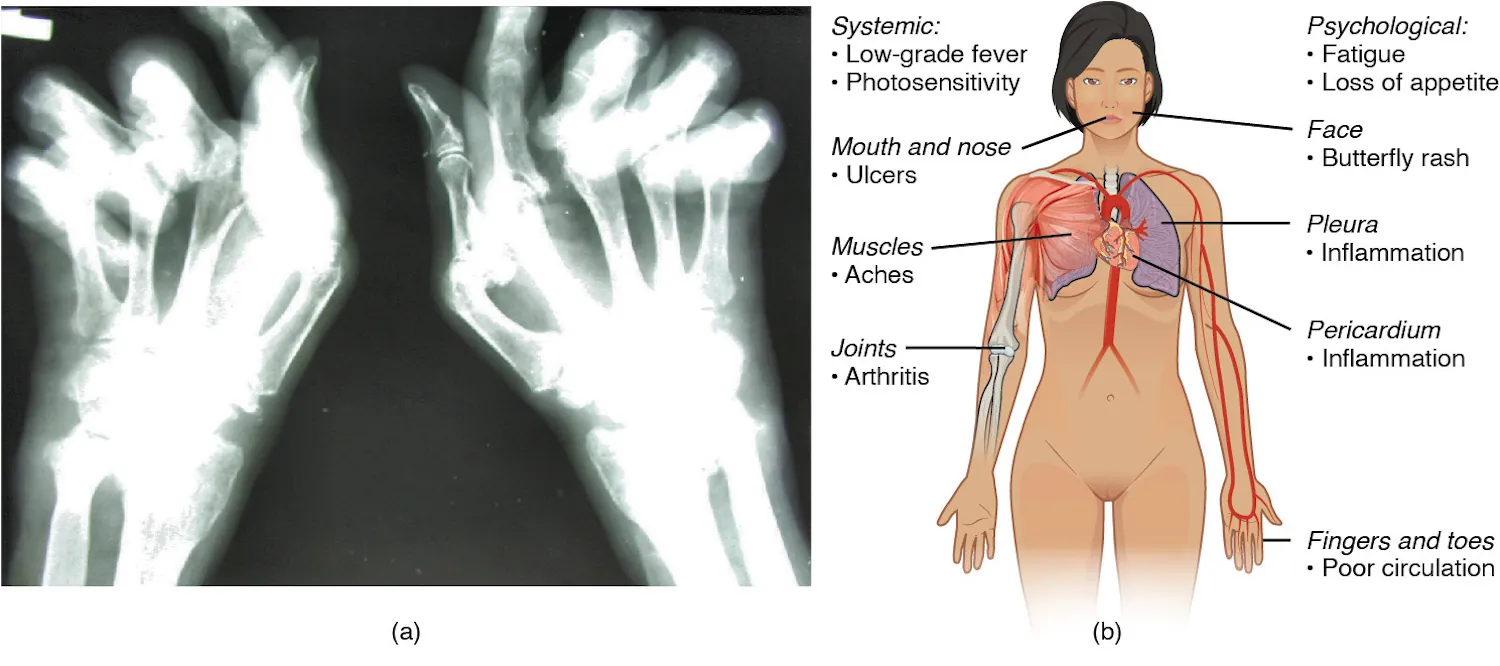
Selected Autoimmune Disease Mechanisms
| Disease | Hypersensitivity Type | Mechanism | Key Features |
|---|---|---|---|
| SLE | III (immune complex), II | Anti-dsDNA/anti-Sm immune complexes deposit in kidneys, joints, skin, serosa | Malar rash, discoid rash, oral ulcers, arthritis, serositis, nephritis (wire-loop lesion), cytopenias; “full house” immunofluorescence (IgG, IgM, IgA, C3, C1q); photosensitivity |
| Rheumatoid arthritis | III, IV | Immune complexes (RF + IgG) in joints; Th1/Th17-mediated synovitis; pannus formation | Symmetric polyarthritis (MCP, PIP, wrist); rheumatoid nodules; swan-neck/boutonniere deformities; anti-CCP most specific |
| Type 1 diabetes | IV | CD8+ T cells destroy pancreatic β-cells; anti-GAD65, anti-insulin, anti-IA2 antibodies | HLA-DR3/DR4; insulitis on biopsy; presents with DKA in young patients |
| Multiple sclerosis | IV | Th1 and Th17 cells attack myelin in CNS; Treg dysfunction | HLA-DR2; oligoclonal bands in CSF; periventricular plaques on MRI; relapsing-remitting course most common |
| Graves disease | II (stimulatory) | Anti-TSH receptor antibodies (thyroid-stimulating immunoglobulins) activate TSH receptor | Hyperthyroidism, diffuse goiter, exophthalmos, pretibial myxedema |
| Myasthenia gravis | II (blocking) | Anti-AChR antibodies block and destroy AChR at NMJ; complement-mediated damage | Fatigable muscle weakness (ptosis, diplopia, dysphagia); thymic hyperplasia/thymoma; improves with AChE inhibitors (edrophonium, pyridostigmine) |
| Celiac disease | IV (and II) | Gluten (gliadin) peptides presented on HLA-DQ2/DQ8 activate Th1 cells; anti-tTG cross-links gliadin to tissue transglutaminase | Villous atrophy, crypt hyperplasia, intraepithelial lymphocytes; dermatitis herpetiformis; IgA anti-tTG and anti-endomysial antibodies |
Caused by hydralazine, procainamide, isoniazid, phenytoin, minocycline, and others. Presents with arthritis, serositis, rash, and positive anti-histone antibodies (present in >95%). Unlike true SLE: renal and CNS involvement are rare, anti-dsDNA is usually negative, complement levels are normal, and symptoms resolve after drug discontinuation.
23 Primary Immunodeficiency Syndromes
Primary immunodeficiencies are inherited defects in immune system components. They are broadly classified by the arm of immunity affected.
B-Cell (Humoral) Deficiencies
| Disorder | Defect | Presentation | Diagnosis |
|---|---|---|---|
| X-linked (Bruton) agammaglobulinemia | BTK (Bruton tyrosine kinase) mutation; no B-cell maturation past pre-B stage | Males; recurrent sinopulmonary infections after 6 months (maternal IgG wanes); absent lymph node germinal centers | Absent B cells, absent all Ig classes; no tonsils/adenoids |
| Common variable immunodeficiency (CVID) | Defective B-cell differentiation to plasma cells; heterogeneous genetics | Most common symptomatic primary immunodeficiency in adults; recurrent sinopulmonary infections; increased risk of autoimmunity and lymphoma | Low IgG (and often IgA); normal B-cell count; poor vaccine responses |
| Selective IgA deficiency | Failure of IgA-producing B cells | Most common primary immunodeficiency overall; usually asymptomatic; recurrent mucosal infections; anaphylaxis risk with IgA-containing blood products | IgA <7 mg/dL; normal IgG and IgM |
T-Cell Deficiencies
| Disorder | Defect | Presentation |
|---|---|---|
| DiGeorge syndrome | 22q11.2 deletion → failure of 3rd/4th pharyngeal pouch development → thymic aplasia, parathyroid hypoplasia | T-cell deficiency (variable severity), hypocalcemia (tetany), cardiac defects (truncus arteriosus, tetralogy of Fallot), facial abnormalities; no thymic shadow on CXR |
| IL-12 receptor deficiency | Defective Th1 response (no IFN-γ production) | Disseminated mycobacterial and fungal infections |
Combined B & T Cell Deficiencies
| Disorder | Defect | Presentation |
|---|---|---|
| SCID (Severe Combined Immunodeficiency) | Multiple genetic causes: X-linked (IL-2R γ-chain/IL-7R defect, most common), ADA deficiency, RAG1/2 deficiency | Failure to thrive, chronic diarrhea, opportunistic infections (PCP, CMV, Candida) within first months of life; absent thymic shadow; lymphopenia; fatal without bone marrow transplant or gene therapy |
| Hyper-IgM syndrome | X-linked: CD40L deficiency; AR: AID deficiency | Elevated IgM, very low IgG/IgA/IgE; recurrent pyogenic and opportunistic infections (Pneumocystis, Cryptosporidium) |
| Wiskott–Aldrich syndrome | WASp gene mutation (X-linked); defective actin cytoskeleton reorganization | Triad: thrombocytopenia (small platelets), eczema, recurrent infections; increased risk of lymphoma and autoimmunity |
| Ataxia-telangiectasia | ATM gene mutation; defective DNA repair | Cerebellar ataxia, oculocutaneous telangiectasias, IgA deficiency, elevated AFP, increased lymphoma/leukemia risk; sensitivity to ionizing radiation |
Phagocyte Deficiencies
| Disorder | Defect | Presentation |
|---|---|---|
| Chronic Granulomatous Disease (CGD) | NADPH oxidase deficiency (X-linked or AR) | Recurrent infections with catalase-positive organisms (S. aureus, Aspergillus, Serratia, Nocardia, Burkholderia cepacia); granuloma formation; negative NBT/DHR test |
| Leukocyte Adhesion Deficiency Type 1 | CD18 (integrin β2) deficiency; leukocytes cannot adhere/transmigrate | Delayed umbilical cord separation (>30 days), recurrent bacterial infections without pus formation, marked leukocytosis (neutrophils cannot leave blood) |
| Chédiak–Higashi syndrome | LYST gene; defective lysosomal trafficking | Giant granules in neutrophils, partial albinism, peripheral neuropathy, recurrent pyogenic infections |
B-cell/humoral defects → encapsulated bacteria (S. pneumoniae, H. influenzae), Giardia. T-cell defects → intracellular organisms (viruses, fungi, mycobacteria, Pneumocystis). Phagocyte defects → catalase-positive bacteria, fungi. Complement defects → Neisseria (terminal), SLE-like (early classical).
Additional Immunodeficiency Syndromes
| Disorder | Defect | Key Features |
|---|---|---|
| Hyper-IgE syndrome (Job syndrome) | STAT3 mutation (AD) or DOCK8 mutation (AR) | Markedly elevated IgE (>2,000 IU/mL); recurrent staphylococcal abscesses (“cold” abscesses — lack warmth and erythema due to impaired neutrophil chemotaxis); eczema; retained primary teeth; characteristic facies; pathologic fractures; STAT3 type has no increased viral infections; DOCK8 type has severe viral skin infections |
| Bare lymphocyte syndrome Type II | MHC II transcription factor deficiency (CIITA, RFX5, RFXAP, RFXANK) | No MHC II expression → no CD4+ T-cell positive selection → SCID-like phenotype; severe infections in infancy |
| Complement deficiency (C2) | Most common complement deficiency overall | SLE-like syndrome; C2 deficiency impairs classical pathway immune complex clearance |
| Myeloperoxidase deficiency | MPO enzyme deficiency in neutrophils | Most common neutrophil enzyme deficiency; usually clinically silent; mild increase in Candida infections in diabetic patients; positive DHR test (NADPH oxidase intact) |
| IL-12 / IFN-γ axis defects | IL-12, IL-12R, IFN-γR, or STAT1 mutations | Susceptibility to mycobacterial infections (both TB and NTM); disseminated BCG infection after vaccination |
24 Acquired Immunodeficiency (HIV/AIDS)
Human Immunodeficiency Virus (HIV) is a lentivirus (retrovirus) that primarily infects CD4+ T cells, macrophages, and dendritic cells via the CD4 receptor and a co-receptor (CCR5 in early infection, CXCR4 in late infection). It carries reverse transcriptase, integrase, and protease — key drug targets.
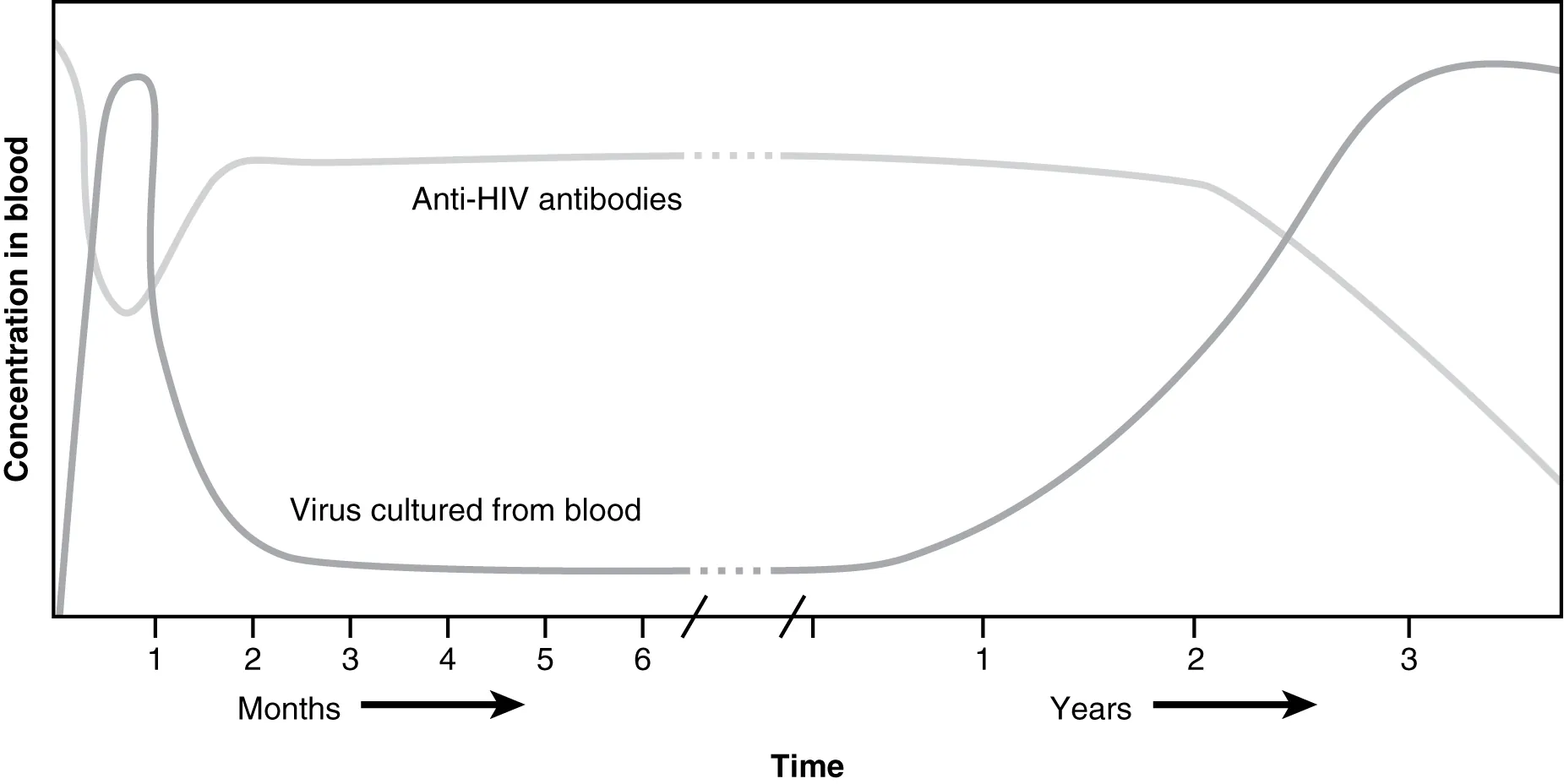
HIV Immunopathogenesis
HIV causes progressive immune destruction through multiple mechanisms:
- Direct cytopathic effect: Viral replication and budding kills infected CD4+ T cells
- Syncytia formation: gp120/gp41 on infected cells fuses with CD4/CXCR4 on uninfected cells → multinucleated giant cells → death
- Bystander apoptosis: Chronic immune activation causes apoptosis of uninfected CD4+ T cells
- Immune exhaustion: Persistent antigen stimulation leads to T-cell exhaustion (PD-1, CTLA-4 upregulation)
- GALT depletion: Massive early CD4+ T-cell loss in gut mucosa → increased microbial translocation → chronic systemic immune activation
Natural History
| Stage | Timing | Features |
|---|---|---|
| Acute retroviral syndrome | 2–4 weeks post-infection | Mono-like illness (fever, pharyngitis, lymphadenopathy, rash, myalgias); high viral load; p24 antigen positive; antibody may be negative (window period) |
| Clinical latency | Years (median ~10 years without treatment) | Asymptomatic or generalized lymphadenopathy; gradual CD4 decline (~50–80 cells/μL/year); virus replicating in lymph nodes |
| AIDS | CD4 <200 or AIDS-defining illness | Opportunistic infections, malignancies (Kaposi sarcoma, primary CNS lymphoma, cervical cancer), wasting |
Opportunistic Infections by CD4 Count
| CD4 Count | Opportunistic Infections |
|---|---|
| <500 | Oral thrush (Candida), hairy leukoplakia (EBV), Kaposi sarcoma (HHV-8), TB reactivation |
| <200 | Pneumocystis jirovecii pneumonia (PCP), disseminated histoplasmosis/coccidioidomycosis |
| <100 | Toxoplasma encephalitis, Cryptococcal meningitis, esophageal candidiasis |
| <50 | CMV retinitis, disseminated MAC, primary CNS lymphoma (EBV), progressive multifocal leukoencephalopathy (JC virus) |
HIV Life Cycle & Drug Targets
| Step | Mechanism | Drug Class (Target) |
|---|---|---|
| 1. Attachment & entry | gp120 binds CD4, then gp41 mediates fusion via co-receptor (CCR5 or CXCR4) | Maraviroc (CCR5 antagonist); enfuvirtide (fusion inhibitor, binds gp41) |
| 2. Reverse transcription | Viral RNA → dsDNA by reverse transcriptase | NRTIs (tenofovir, emtricitabine, zidovudine, lamivudine); NNRTIs (efavirenz, rilpivirine) |
| 3. Integration | Viral DNA integrates into host genome via integrase | INSTIs (dolutegravir, raltegravir, bictegravir) — preferred first-line agents |
| 4. Transcription & translation | Host machinery transcribes viral genes, translates polyproteins | No current drug targets at this step |
| 5. Assembly & maturation | Protease cleaves polyproteins into functional viral proteins | PIs (ritonavir, darunavir, atazanavir); ritonavir used as pharmacokinetic booster (CYP3A4 inhibitor) |
| 6. Budding | New virions bud from host cell membrane | No current drug targets at this step |
Diagnosis
Fourth-generation HIV test detects both p24 antigen and HIV-1/2 antibodies (best initial test). Confirmatory: HIV-1/2 antibody differentiation assay. If antibody indeterminate: HIV-1 RNA (viral load) PCR. In neonates (maternal antibodies present): use HIV DNA PCR or RNA PCR (not antibody tests).
Treatment & Prevention
Standard ART regimen: 2 NRTIs (backbone) + 1 INSTI (preferred third agent). Goal: undetectable viral load (<50 copies/mL). PrEP (pre-exposure prophylaxis): tenofovir/emtricitabine or cabotegravir for high-risk individuals. PEP (post-exposure prophylaxis): 3-drug ART within 72 hours of exposure for 28 days.
25 Transplant Immunology & Rejection
Types of Grafts
| Graft Type | Definition | Example |
|---|---|---|
| Autograft | Self to self | Skin graft from thigh to face; no rejection |
| Isograft (syngeneic) | Identical twin to twin | No rejection (genetically identical) |
| Allograft | Same species, different individual | Cadaveric kidney transplant; most common clinical scenario |
| Xenograft | Different species | Pig heart valve in human; high rejection risk |
Rejection Types
| Type | Timing | Mechanism | Pathology | Treatment |
|---|---|---|---|---|
| Hyperacute | Minutes–hours | Preformed recipient antibodies against donor HLA or ABO antigens → complement activation → vascular thrombosis | Widespread thrombosis, graft infarction | Prevention only (crossmatch testing); graft must be removed |
| Acute cellular | Weeks–months | Recipient CD4+ and CD8+ T cells attack donor MHC antigens | Lymphocytic infiltration of graft (tubulitis in kidney) | Increase immunosuppression (pulse steroids, anti-thymocyte globulin) |
| Acute humoral (antibody-mediated) | Weeks–months | Recipient antibodies against donor HLA → complement activation in graft vessels | C4d deposition in peritubular capillaries; neutrophilic infiltrate | Plasmapheresis, IVIg, rituximab |
| Chronic | Months–years | Both antibody and cell-mediated; vascular intimal fibrosis (transplant vasculopathy) | Fibrosis, vascular thickening (“transplant vasculopathy”); gradual graft dysfunction | Retransplantation (irreversible) |
Graft-versus-Host Disease (GvHD)
Occurs when donor T cells in the graft attack immunocompromised host tissues. Most common after bone marrow/stem cell transplant (also after non-irradiated blood transfusion to immunocompromised patient). Targets: skin (maculopapular rash), liver (jaundice, elevated LFTs), GI tract (diarrhea, abdominal pain). Acute GvHD occurs <100 days post-transplant; chronic GvHD occurs >100 days and resembles systemic sclerosis.
Immunosuppressive Agents
| Drug | Mechanism | Key Side Effects |
|---|---|---|
| Cyclosporine | Calcineurin inhibitor (binds cyclophilin) → blocks IL-2 transcription in T cells | Nephrotoxicity, hypertension, gingival hyperplasia, hirsutism, tremor |
| Tacrolimus (FK506) | Calcineurin inhibitor (binds FKBP) → blocks IL-2 transcription | Nephrotoxicity, diabetes, neurotoxicity (more potent than cyclosporine) |
| Sirolimus (rapamycin) | mTOR inhibitor (binds FKBP) → blocks T-cell proliferation at G1→S | Hyperlipidemia, myelosuppression, poor wound healing; NOT nephrotoxic |
| Mycophenolate mofetil | Inosine monophosphate dehydrogenase inhibitor → blocks de novo purine synthesis in lymphocytes | GI upset, myelosuppression, teratogenicity |
| Azathioprine | Purine analog → inhibits DNA synthesis (prodrug → 6-mercaptopurine) | Myelosuppression; metabolized by TPMT (check before dosing); drug interaction with allopurinol |
| Basiliximab | Anti-IL-2R (CD25) monoclonal antibody → blocks T-cell proliferation | Generally well tolerated; used as induction therapy |
| Anti-thymocyte globulin (ATG) | Polyclonal antibodies against T cells → lymphocyte depletion | Cytokine release syndrome, serum sickness, myelosuppression |
Immunosuppression Protocols
Transplant immunosuppression typically uses a three-drug regimen:
- Induction: Anti-thymocyte globulin (ATG) or basiliximab (anti-IL-2R) given peri-operatively for intense early immunosuppression
- Maintenance: Triple therapy — calcineurin inhibitor (tacrolimus preferred) + antimetabolite (mycophenolate) + low-dose corticosteroids
- Rejection treatment: Pulse IV methylprednisolone for mild acute rejection; ATG or OKT3 for steroid-resistant rejection; plasmapheresis + IVIg + rituximab for antibody-mediated rejection
GvHD prophylaxis in bone marrow transplant: tacrolimus or cyclosporine + methotrexate or mycophenolate. T-cell depletion of the graft reduces GvHD but increases relapse risk (loss of beneficial graft-versus-leukemia/GVL effect). Acute GvHD treatment: corticosteroids first-line; ruxolitinib (JAK inhibitor) for steroid-refractory cases. The graft-versus-leukemia (GVL) effect is a beneficial consequence of donor T cells recognizing and killing residual leukemia cells — this is why complete T-cell depletion is not always desirable.
26 Tumor Immunology & Immunotherapy
Immune Surveillance
The immune system continuously monitors for and eliminates transformed cells via the cancer immunoediting model: (1) Elimination — immune cells (NK, CTLs) kill nascent tumor cells; (2) Equilibrium — immune pressure sculpts tumor variants; (3) Escape — tumor evades immunity through downregulation of MHC I, secretion of immunosuppressive cytokines (TGF-β, IL-10), PD-L1 expression, and recruitment of Tregs/MDSCs.
Tumor Immune Evasion Mechanisms
| Mechanism | Effect |
|---|---|
| MHC I downregulation | Evades CD8+ T-cell recognition (but becomes NK cell target) |
| PD-L1 overexpression | Engages PD-1 on T cells → T-cell exhaustion/anergy |
| Treg/MDSC recruitment | Creates immunosuppressive tumor microenvironment |
| Antigen loss / neoantigen masking | Tumor variants lacking immunogenic antigens are selected |
| FasL expression on tumor | Induces apoptosis of Fas-expressing infiltrating T cells |
Cancer Immunotherapy Approaches
| Approach | Examples | Mechanism |
|---|---|---|
| Checkpoint inhibitors | Pembrolizumab, nivolumab (anti-PD-1); atezolizumab (anti-PD-L1); ipilimumab (anti-CTLA-4) | Block inhibitory signals, restoring T-cell anti-tumor activity |
| CAR-T cell therapy | Tisagenlecleucel (anti-CD19), axicabtagene ciloleucel | Patient’s T cells engineered to express chimeric antigen receptor targeting tumor antigen (e.g., CD19 in B-ALL); cytokine release syndrome is major adverse effect |
| Monoclonal antibodies | Rituximab (anti-CD20), trastuzumab (anti-HER2), cetuximab (anti-EGFR) | ADCC, complement-mediated lysis, receptor blockade |
| Cancer vaccines | Sipuleucel-T (prostate cancer), HPV vaccine (prevention) | Stimulate anti-tumor immune response |
| Cytokine therapy | High-dose IL-2 (melanoma, RCC), IFN-α (melanoma, hairy cell leukemia) | Boost T-cell and NK cell anti-tumor activity |
| Bispecific antibodies | Blinatumomab (anti-CD19/CD3) | Bridge T cells to tumor cells, facilitating direct killing |
Tumor Antigens
| Antigen Type | Description | Examples |
|---|---|---|
| Tumor-specific antigens (TSA) | Unique to tumor cells (neoantigens from mutations); ideal immunotherapy targets | Mutated p53, mutated RAS, BCR-ABL fusion protein |
| Tumor-associated antigens (TAA) | Overexpressed or aberrantly expressed normal proteins; shared with normal tissues | HER2/neu (breast), PSA (prostate), AFP (α-fetoprotein, HCC/germ cell), CEA (colorectal), CA-125 (ovarian) |
| Cancer-testis antigens | Normally expressed only in testes (immune-privileged site); re-expressed in tumors | MAGE, NY-ESO-1 |
| Oncoviral antigens | Viral proteins in virus-associated cancers | EBV antigens (Burkitt lymphoma, NPC), HPV E6/E7 (cervical cancer), HHV-8 (Kaposi sarcoma) |
27 Vaccination & Immunoprophylaxis
Vaccine Types
| Type | Mechanism | Examples | Key Points |
|---|---|---|---|
| Live attenuated | Weakened pathogen replicates, inducing strong humoral + cellular immunity | MMR, varicella, rotavirus, BCG, oral polio (Sabin), yellow fever, intranasal influenza (FluMist) | Strongest immune response; contraindicated in immunocompromised and pregnant patients; risk of reversion to virulence (oral polio) |
| Inactivated (killed) | Killed pathogen; induces humoral immunity (weaker, needs boosters) | Influenza (injection), hepatitis A, rabies, IPV (Salk polio) | Safe in immunocompromised; weaker immunity requires multiple doses |
| Subunit / recombinant | Purified antigen component | Hepatitis B (HBsAg), HPV (L1 capsid protein), acellular pertussis, influenza (recombinant) | Cannot cause disease; requires adjuvant for adequate response |
| Toxoid | Inactivated toxin | Tetanus, diphtheria | Induces anti-toxin antibodies; requires boosters |
| Conjugate | Polysaccharide linked to protein carrier | PCV13 (pneumococcal), Hib, MenACWY | Converts T-independent to T-dependent response; effective in children <2 years |
| Polysaccharide | Purified capsular polysaccharide | PPSV23 (pneumococcal), MenB | T-independent response (IgM only, no memory); ineffective in <2 years |
| mRNA | mRNA encoding antigen delivered in lipid nanoparticles; translated by host cells | COVID-19 (Pfizer-BioNTech, Moderna) | Rapid development platform; strong humoral and cellular response; requires cold storage |
| Viral vector | Adenovirus vector carries gene encoding target antigen | COVID-19 (J&J/Janssen), Ebola vaccine | Pre-existing vector immunity may reduce efficacy |
Key Vaccination Concepts
- Herd immunity: When a sufficient proportion of the population is immune, transmission is interrupted, protecting unvaccinated individuals. Threshold varies by pathogen (measles: ~95%; polio: ~80%; influenza: ~75%).
- Passive immunization: Preformed antibodies given for immediate protection. Examples: anti-D immunoglobulin (RhoGAM), rabies immunoglobulin, hepatitis B immunoglobulin (HBIG), tetanus immunoglobulin, IVIg for Kawasaki disease, palivizumab (anti-RSV) for premature infants.
- Adjuvants: Substances (aluminum salts, MF59, AS01B) that enhance the immune response by activating innate immunity (PAMP mimicry), creating a depot effect, and promoting APC activation.
- Primary vs. secondary immune response: Primary response is slower (7–10 days), produces mainly IgM, lower affinity. Secondary response (booster/re-exposure) is faster (1–3 days), produces mainly IgG, higher affinity (due to memory B cells and affinity maturation). This is the basis for booster vaccinations.
- Immune correlates of protection: The specific immune parameter that confers protection varies by pathogen — neutralizing antibody titer (most vaccines), cell-mediated immunity (TB, some viral infections), or mucosal IgA (enteric pathogens).
Special Vaccination Considerations
| Population | Consideration |
|---|---|
| Immunocompromised patients | No live vaccines; killed/subunit/toxoid vaccines are safe but may have reduced efficacy; check antibody titers post-vaccination |
| Pregnant women | No live vaccines (risk of fetal infection); Tdap recommended at 27–36 weeks each pregnancy (passive antibody transfer to neonate); inactivated influenza and COVID-19 vaccines recommended |
| Asplenic patients | Pneumococcal (PCV20 or PCV15 + PPSV23), meningococcal (MenACWY + MenB), Hib vaccines; administer ≥2 weeks before elective splenectomy if possible |
| Healthcare workers | Hepatitis B (check anti-HBs titer), annual influenza, Tdap booster, varicella immunity, MMR immunity |
| Neonates | Hepatitis B vaccine at birth; maternal antibodies provide passive protection for ~6 months; conjugate vaccines effective after 2 months |
28 High-Yield Review
- MHC I presents endogenous antigen to CD8+ T cells; MHC II presents exogenous antigen to CD4+ T cells (Rule of 8).
- All nucleated cells express MHC I; only professional APCs express MHC II.
- IgG is the only antibody that crosses the placenta; IgA dominates mucosal immunity; IgM is the first responder and best complement activator.
- Type I hypersensitivity = IgE/mast cells (anaphylaxis). Type II = IgG/IgM against cell surface (hemolysis). Type III = immune complexes (serum sickness, SLE nephritis). Type IV = T cells (PPD, contact dermatitis, transplant rejection).
- Complement: Classical (antibody/C1q), Lectin (MBL/mannose), Alternative (C3 tick-over). All converge at C3.
- C3b = opsonization; C3a/C5a = anaphylatoxins; C5a = neutrophil chemotaxis; C5b–C9 = MAC (lysis).
- Terminal complement deficiency (C5–C9) → recurrent Neisseria infections.
- C1-INH deficiency → hereditary angioedema (bradykinin-mediated).
- DAF/CD59 deficiency → PNH (complement-mediated hemolysis; treat with eculizumab, anti-C5).
- Bruton agammaglobulinemia: BTK mutation, no mature B cells, no Ig; X-linked; infections after 6 months.
- DiGeorge: 22q11.2 deletion, thymic aplasia, T-cell deficiency, hypocalcemia, cardiac defects.
- SCID: absent T cells (and often B/NK); most common = X-linked IL-2R γ-chain deficiency; fatal without BMT.
- CGD: NADPH oxidase defect → no oxidative burst → catalase-positive infections; negative NBT/DHR.
- Leukocyte adhesion deficiency: CD18 deficiency → delayed cord separation, no pus, leukocytosis.
- Hyper-IgM: CD40L deficiency (X-linked) → no class switching → elevated IgM, low IgG/IgA/IgE.
- Wiskott–Aldrich: WASp mutation → thrombocytopenia + eczema + infections (X-linked).
- AIRE mutations → APECED (APS-1); FoxP3 mutations → IPEX syndrome (Treg deficiency).
- HIV infects CD4+ T cells via CD4 + CCR5 (early) or CXCR4 (late); AIDS = CD4 <200.
- Hyperacute rejection = preformed antibodies (minutes); Acute = T cells (weeks); Chronic = fibrosis (months–years).
- GvHD: donor T cells attack host; skin, liver, GI; occurs in bone marrow transplant.
- Cyclosporine and tacrolimus = calcineurin inhibitors → block IL-2; sirolimus = mTOR inhibitor (non-nephrotoxic).
- Anti-dsDNA and anti-Smith are specific for SLE; ANA is sensitive but not specific.
- Anti-CCP is most specific for rheumatoid arthritis; RF is sensitive but not specific.
- HLA-B27 → ankylosing spondylitis and seronegative spondyloarthropathies.
- HLA-DQ2/DQ8 → celiac disease.
- Th1 (IFN-γ, macrophage activation); Th2 (IL-4/5/13, IgE, eosinophils); Th17 (IL-17, neutrophils).
- Checkpoint inhibitors: anti-PD-1 (pembrolizumab, nivolumab), anti-CTLA-4 (ipilimumab); irAEs mimic autoimmunity.
- Live vaccines contraindicated in immunocompromised and pregnant patients; conjugate vaccines work in children <2.
- Positive selection (thymic cortex) = MHC restriction; Negative selection (medulla) = self-tolerance.
- Somatic hypermutation (AID) and affinity maturation occur in germinal centers; class switching requires CD40L-CD40 interaction.
Quick-Reference: Immunodeficiency Diagnostic Clues
| Finding | Think |
|---|---|
| Absent B cells, no Ig | Bruton (XLA) |
| No thymic shadow, hypocalcemia | DiGeorge |
| Negative NBT/DHR test | CGD |
| Giant granules in neutrophils | Chédiak–Higashi |
| Delayed cord separation, no pus | LAD type 1 |
| Eczema + thrombocytopenia + infections (male) | Wiskott–Aldrich |
| Ataxia + telangiectasias + elevated AFP | Ataxia-telangiectasia |
| Elevated IgM, low IgG/A/E | Hyper-IgM syndrome |
| Severe infections in first months, lymphopenia | SCID |
| Recurrent sinopulmonary infections in adult, low IgG | CVID |
| Anaphylaxis to blood products, IgA <7 | Selective IgA deficiency |
| Cold abscesses, very high IgE, eczema, retained teeth | Hyper-IgE (Job) syndrome |
| Recurrent Neisseria meningitis | Terminal complement (C5–C9) deficiency |
| Angioedema without urticaria, low C4 | Hereditary angioedema (C1-INH deficiency) |
High-Yield CD Markers
| CD Marker | Cell / Function | Board Association |
|---|---|---|
| CD3 | All mature T cells (TCR complex signaling) | Pan-T-cell marker; OKT3 (muromonab) target |
| CD4 | Helper T cells; MHC II co-receptor | HIV receptor; CD4 count stages AIDS |
| CD8 | Cytotoxic T cells; MHC I co-receptor | Kills virus-infected and tumor cells |
| CD16/CD56 | NK cells | ADCC (CD16 = FcγRIII); CD56 = NK marker |
| CD19/CD20 | B cells | Rituximab (anti-CD20); CD19 = CAR-T target in B-ALL |
| CD21 | B cells (complement receptor CR2) | EBV receptor (EBV tropism for B cells) |
| CD25 | IL-2Rα (activated T cells, Tregs) | Basiliximab target; elevated soluble IL-2R in HLH |
| CD28 | T-cell co-stimulatory receptor (binds B7) | Abatacept (CTLA-4-Ig) blocks this pathway |
| CD40 | APCs, B cells | CD40L deficiency → Hyper-IgM syndrome |
| CD80/86 (B7) | APCs (co-stimulatory ligand) | Binds CD28 (activation) or CTLA-4 (inhibition) |
| CD95 (Fas) | Apoptosis receptor on many cells | Fas mutations → ALPS |
| CD117 (c-Kit) | Mast cells, hematopoietic progenitors | Mastocytosis; imatinib target in c-Kit+ GIST |
Laboratory Tests in Immunology
| Test | What It Measures | Clinical Use |
|---|---|---|
| Flow cytometry (FACS) | Cell surface markers (CD counts) | CD4 count in HIV; T/B/NK cell enumeration in immunodeficiency; leukemia/lymphoma immunophenotyping |
| Serum immunoglobulin levels | IgG, IgA, IgM, IgE quantitation | Hypogammaglobulinemia (Bruton, CVID), elevated IgE (atopy, parasites, Hyper-IgE syndrome) |
| Complement levels (CH50, C3, C4) | Functional complement activity | CH50 = 0 suggests complete deficiency; low C3/C4 in active SLE, PSGN; low C4 with normal C3 in hereditary angioedema |
| NBT / DHR test | Neutrophil oxidative burst | Negative/absent in CGD (NADPH oxidase deficiency) |
| Direct Coombs test (DAT) | Antibodies bound to patient RBCs | Autoimmune hemolytic anemia, hemolytic disease of newborn, drug-induced hemolysis |
| Indirect Coombs test | Free anti-RBC antibodies in serum | Blood bank cross-matching; antibody identification |
| ANA (antinuclear antibody) | Antibodies against nuclear components | Screening for SLE and other connective tissue diseases; patterns (homogeneous, speckled, nucleolar, centromere) suggest specific diagnoses |
| Skin prick test | IgE-mediated reactivity to specific allergens | Wheal-and-flare response within 15–20 minutes confirms Type I hypersensitivity |
| Mixed lymphocyte reaction (MLR) | T-cell reactivity to donor HLA | Pre-transplant assessment of donor-recipient compatibility |
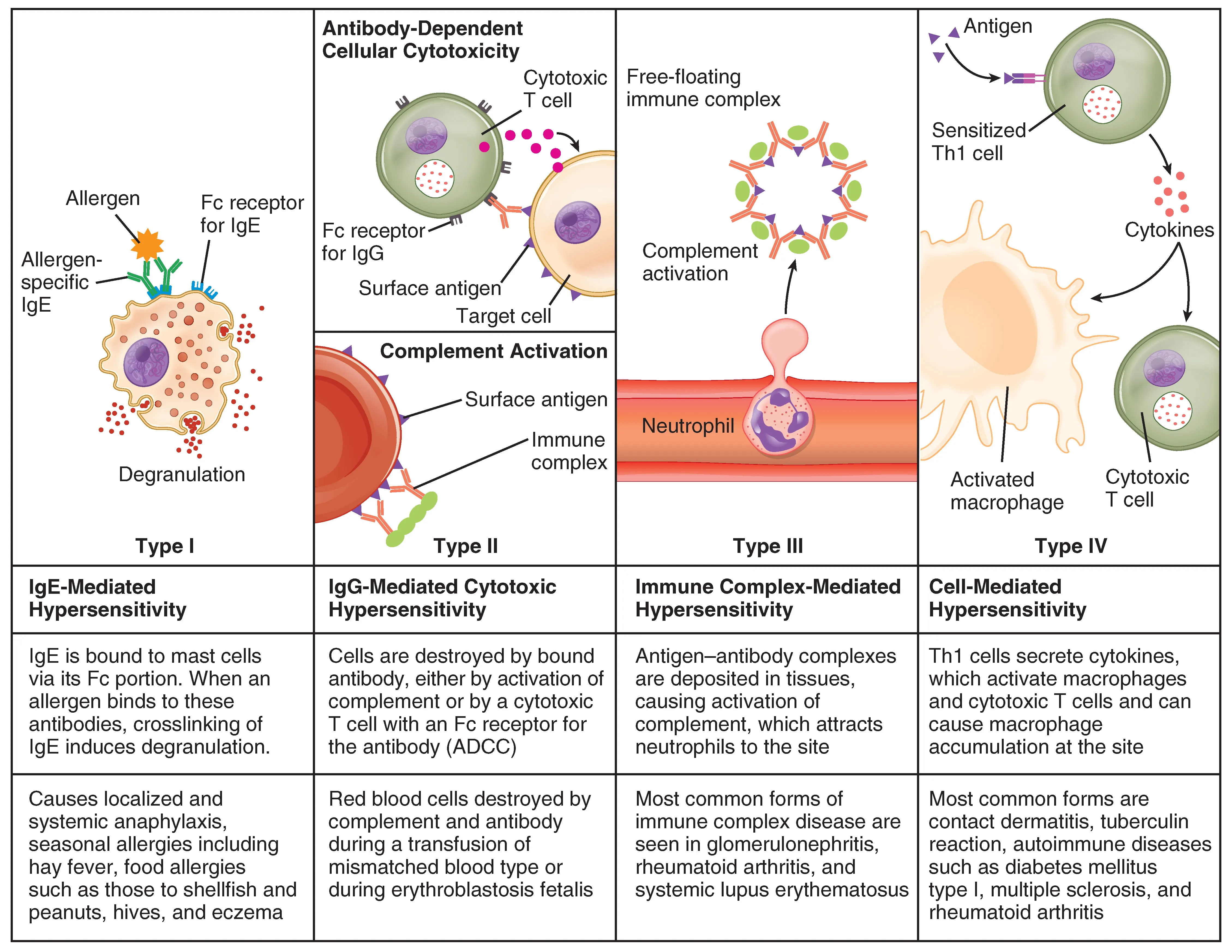
Hypersensitivity Quick-Reference Summary
| Type | Mediator | Timing | Mechanism | Classic Examples |
|---|---|---|---|---|
| I | IgE | Minutes | Mast cell degranulation | Anaphylaxis, asthma, allergic rhinitis |
| II | IgG, IgM | Hours | Cell-surface antibodies → complement, ADCC, receptor dysfunction | AIHA, Goodpasture, Graves, MG |
| III | IgG (immune complexes) | Hours–days | Complex deposition → complement → neutrophilic inflammation | SLE nephritis, serum sickness, PSGN |
| IV | T cells | 24–72 hours | Th1/macrophage activation or CD8+ cytotoxicity | PPD, contact dermatitis, graft rejection, TB granulomas |