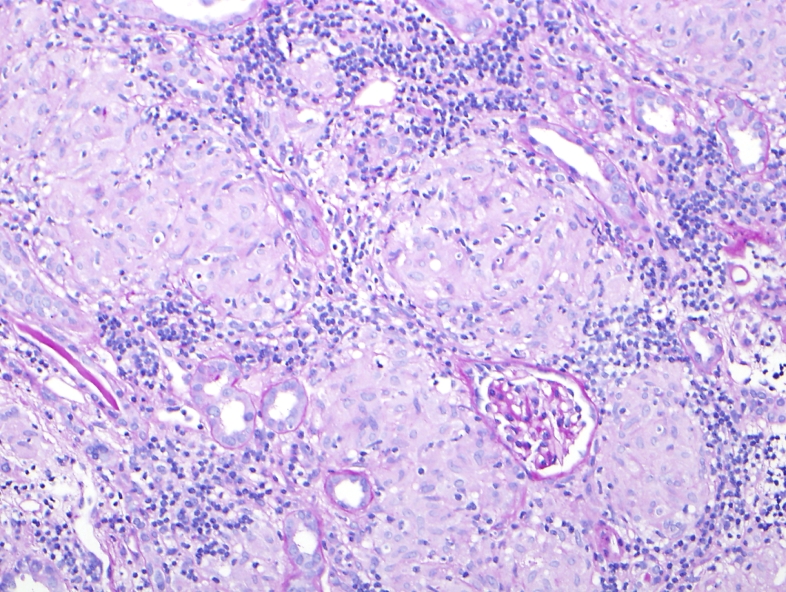
Cellular injury, inflammation, hemodynamic disorders, neoplasia, immunopathology, genetic disease mechanisms, and every pathophysiologic process, mediator, and disease mechanism across the full scope of general pathology.
01 Overview & Scope of Pathophysiology
Pathophysiology is the study of disordered physiological processes that underlie disease. It bridges the gap between basic science and clinical medicine by explaining how and why diseases develop, progress, and produce clinical manifestations. General pathology encompasses the core mechanisms — cell injury, inflammation, hemodynamic derangements, neoplasia, and immune dysfunction — that recur across virtually every organ system and clinical specialty.
WHY PATHOPHYSIOLOGY MATTERS
Every clinical sign, laboratory abnormality, and imaging finding reflects an underlying pathophysiologic mechanism. A clinician who understands the “why” behind disease can predict complications, interpret atypical presentations, choose rational therapies, and avoid diagnostic pitfalls that rote memorization alone cannot address.
The Four Pillars of Pathology
Pillar
Focus
Key Questions
Etiology
Cause of disease
What agent or defect initiates the process?
Pathogenesis
Mechanism of disease development
What sequence of events leads from cause to lesion?
Morphologic Changes
Structural alterations in cells/tissues
What do gross and microscopic changes look like?
Clinical Significance
Functional consequences
What symptoms, signs, and lab findings result?
When evaluating any disease, always organize your thinking around etiology → pathogenesis → morphology → clinical manifestation. This framework applies universally from myocardial infarction to systemic lupus to colon cancer.
Systemic inflammation triggers the liver to produce acute phase proteins under the influence of IL-6, IL-1, and TNF-α. These proteins serve as important clinical markers and mediators of the inflammatory response.
Positive Acute Phase Reactants (↑)
Function / Clinical Use
C-reactive protein (CRP)
Opsonin (binds phosphocholine on bacteria); activates complement; most widely used clinical marker of inflammation; rises within 6 hours
Fibrinogen
Coagulation factor I; elevates ESR (promotes rouleaux formation); contributes to hypercoagulable state in inflammation
Ferritin
Iron storage protein; sequesters iron from pathogens; very high in adult-onset Still disease and hemophagocytic lymphohistiocytosis (HLH)
Hepcidin
Master regulator of iron; blocks ferroportin → traps iron in macrophages and enterocytes; mediates anemia of chronic disease
Serum amyloid A (SAA)
Precursor of AA amyloid in chronic inflammation; lipoprotein associated
Complement (C3, C4)
Enhanced complement activation during inflammation
Negative Acute Phase Reactants (↓)
Significance
Albumin
Decreased hepatic synthesis during inflammation; shifts to producing positive APRs; hypoalbuminemia contributes to edema
Transferrin
Decreased iron transport capacity; contributes to functional iron deficiency in chronic disease
Transthyretin (prealbumin)
Short half-life (~2 days) makes it a sensitive marker of nutritional status; drops rapidly in inflammation
The ESR (erythrocyte sedimentation rate) is elevated in inflammation primarily because increased fibrinogen promotes RBC rouleaux formation, causing faster sedimentation. ESR is not a direct measure of inflammation but rather reflects the protein milieu. CRP is more specific and responsive (rises and falls faster). ESR is disproportionately elevated in multiple myeloma and Waldenström macroglobulinemia due to high immunoglobulin levels promoting rouleaux.
02 Cellular Homeostasis & Adaptation
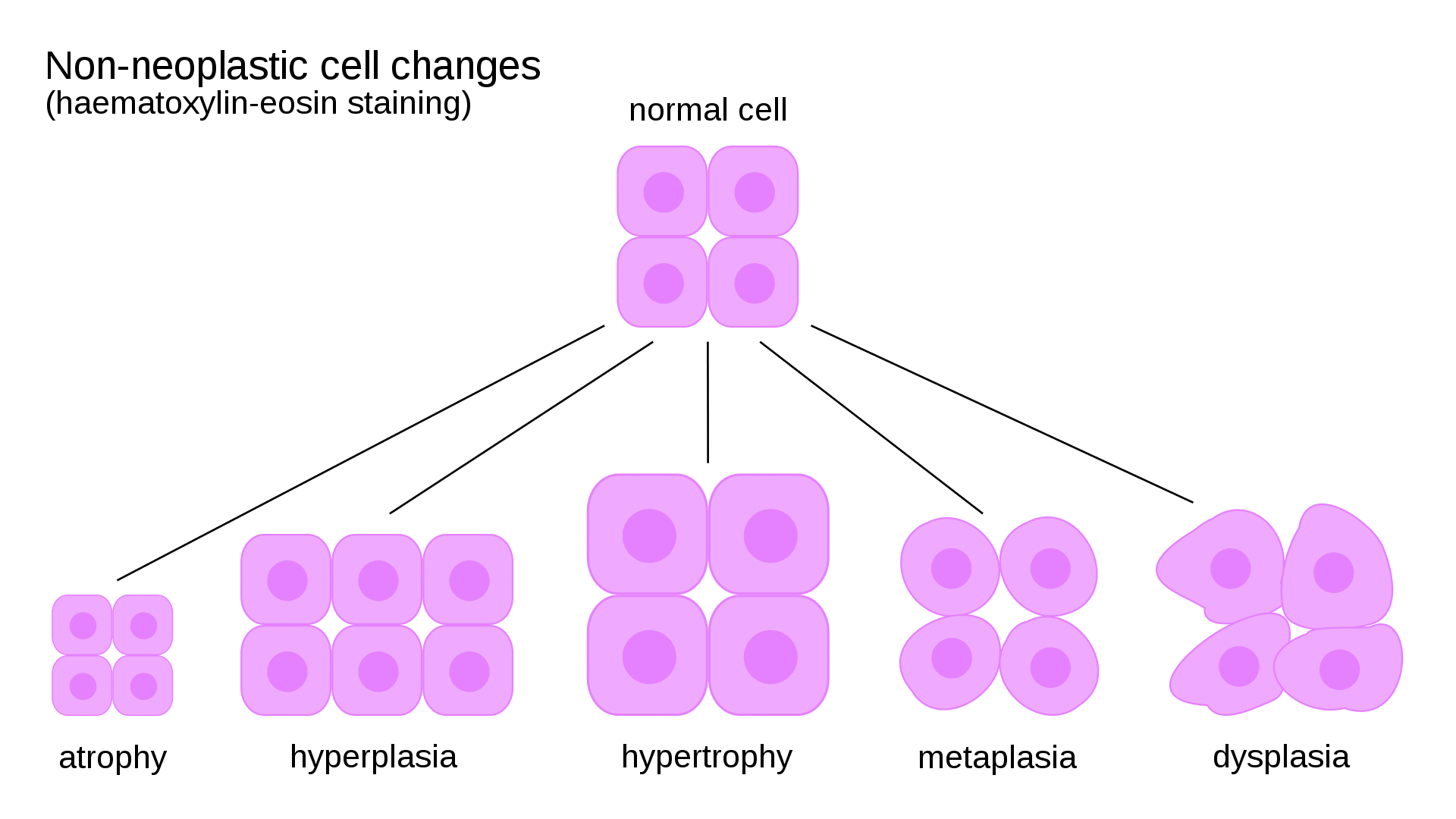
Normal cells operate within a narrow range of structure and function defined by their genetic program, metabolic demands, and extracellular signals. When stressed, cells can adapt through reversible changes in size, number, or phenotype. Adaptation is a key concept distinguishing reversible from irreversible cell injury.
Left ventricular hypertrophy in hypertension; skeletal muscle hypertrophy with exercise
Hyperplasia
Increase in cell number
Growth factor–driven cell proliferation
Endometrial hyperplasia from excess estrogen; compensatory liver regeneration after partial hepatectomy
Atrophy
Decrease in cell size and organelle content
Decreased protein synthesis + increased degradation (ubiquitin-proteasome pathway, autophagy)
Disuse atrophy of immobilized limb; denervation atrophy; senile atrophy of brain
Metaplasia
Replacement of one differentiated cell type by another
Reprogramming of stem cells by cytokines, growth factors, extracellular matrix
Squamous metaplasia of bronchial epithelium in smokers; Barrett esophagus (squamous → columnar)
Dysplasia
Disordered growth with loss of uniformity and architectural orientation
Accumulated genetic alterations in proliferating cells
Cervical dysplasia (CIN); colonic dysplasia in ulcerative colitis
CLINICAL CORRELATION
Barrett esophagus is the prototypical example of metaplasia → dysplasia → carcinoma sequence. Chronic GERD causes squamous-to-columnar metaplasia of the distal esophagus, which can progress through low-grade and high-grade dysplasia to esophageal adenocarcinoma. This progression underscores why metaplasia and dysplasia are considered precancerous conditions requiring surveillance.
Figure 1 — Cellular Adaptations. Non-neoplastic changes a cell can undergo in response to stress, including hypertrophy (increased cell size), hyperplasia (increased cell number), atrophy (decreased cell size), and metaplasia (change from one cell type to another).
Hypertrophy and hyperplasia often coexist. The gravid uterus undergoes both smooth muscle hypertrophy (estrogen-driven increase in cell size) and hyperplasia (estrogen-driven increase in cell number). Only cells capable of division can undergo hyperplasia — cardiac myocytes and neurons primarily undergo hypertrophy alone.
03 Key Terminology & Abbreviations
Abbreviation
Full Term
ROS
Reactive oxygen species
TNF
Tumor necrosis factor
IL
Interleukin
NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
COX
Cyclooxygenase
LOX
Lipoxygenase
PG
Prostaglandin
LT
Leukotriene
NO
Nitric oxide
VEGF
Vascular endothelial growth factor
TGF-β
Transforming growth factor beta
DIC
Disseminated intravascular coagulation
DVT
Deep vein thrombosis
PE
Pulmonary embolism
MI
Myocardial infarction
MHC
Major histocompatibility complex
HLA
Human leukocyte antigen
DAMP
Damage-associated molecular pattern
PAMP
Pathogen-associated molecular pattern
TLR
Toll-like receptor
MAC
Membrane attack complex (C5b-9)
Rb
Retinoblastoma protein (tumor suppressor)
BRCA
Breast cancer susceptibility gene
AFP
Alpha-fetoprotein
CEA
Carcinoembryonic antigen
PSA
Prostate-specific antigen
04 Mechanisms of Cell Injury
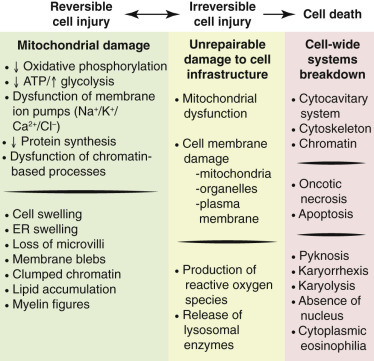
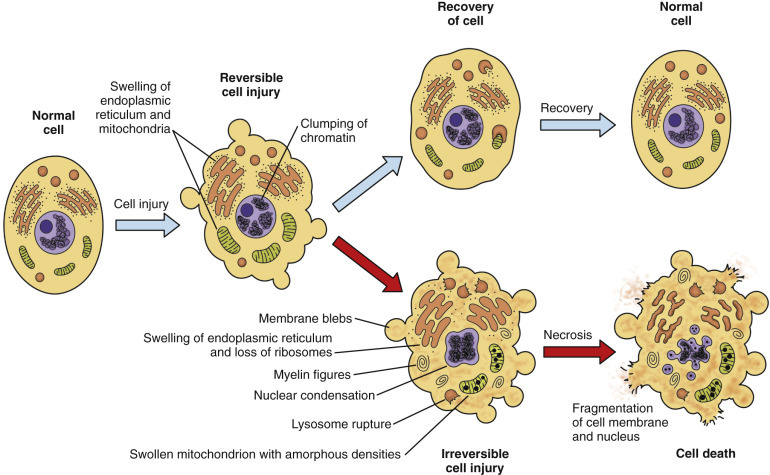
Cell injury occurs when stresses exceed the cell’s ability to adapt. Injury is initially reversible — the cell can return to normal if the stimulus is removed — but persistent or severe stress crosses a “point of no return” leading to irreversible injury and cell death.
Major Causes of Cell Injury
Cause
Mechanism
Examples
Hypoxia / Ischemia
Decreased O2 delivery → impaired oxidative phosphorylation → ATP depletion
Atherosclerotic coronary occlusion (MI); anemia; CO poisoning; respiratory failure
Toxins
Direct damage to membranes, enzymes, or DNA; or generation of toxic metabolites
CCl4 (hepatotoxicity via free radical P-450 metabolism); acetaminophen (NAPQI); ethanol
Infectious agents
Direct cytopathic effect, exotoxins, endotoxins, immune-mediated damage
Scurvy (vitamin C deficiency); kwashiorkor (protein deficiency); obesity
Figure 2 — Ischemic Cell Injury Pathway. Schematic showing the sequence of events in ischemic cell injury, from initial ATP depletion and reversible changes through the point of no return to irreversible injury and cell death.
Sequence of Ischemic Cell Injury
ISCHEMIA → CELL DEATH TIMELINE
Seconds: Cessation of oxidative phosphorylation; ATP begins to fall
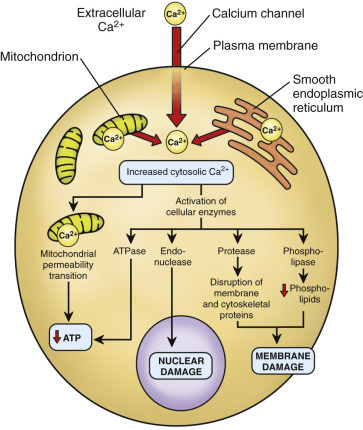
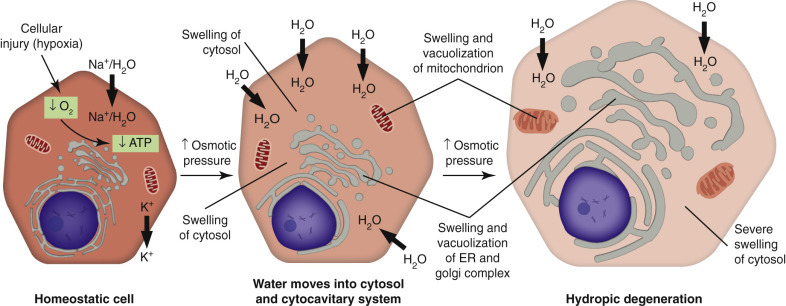
Figure 4 — Cell Injury, Necrosis, and Apoptosis. Overview of the major pathways of cell injury, illustrating how different insults lead to either necrosis (with inflammation) or apoptosis (without inflammation) depending on the nature and severity of the stimulus.Figure 5 — Calcium in Cell Injury. The role of increased cytosolic calcium in mediating cell injury. Massive calcium influx activates destructive enzymes including phospholipases, proteases, endonucleases, and ATPases, contributing to irreversible damage.Figure 6 — Acute Cell Swelling. Histologic and ultrastructural features of acute cell swelling (hydropic change), the earliest and most common manifestation of reversible cell injury caused by failure of the Na+/K+-ATPase pump.
The two morphologic hallmarks of irreversible cell injury are (1) mitochondrial dense amorphous densities (flocculent densities on EM) and (2) plasma membrane disruption. These distinguish the “point of no return” from reversible changes such as cellular swelling and fatty change.
05 Free Radical & Oxidative Injury
Free radicals are chemical species with a single unpaired electron in an outer orbital, making them highly reactive. Reactive oxygen species (ROS) are the most important free radicals in biological systems and play a central role in cell injury, aging, and cancer.
Major Reactive Oxygen Species
Species
Symbol
Source
Superoxide anion
O2•−
Mitochondrial electron transport chain leak; NADPH oxidase (phagocytes); xanthine oxidase
Lipid-soluble chain-breaking antioxidant in membranes
Vitamin C (ascorbate)
Water-soluble antioxidant; regenerates vitamin E
Ferritin & ceruloplasmin
Sequester free iron and copper, preventing Fenton reaction
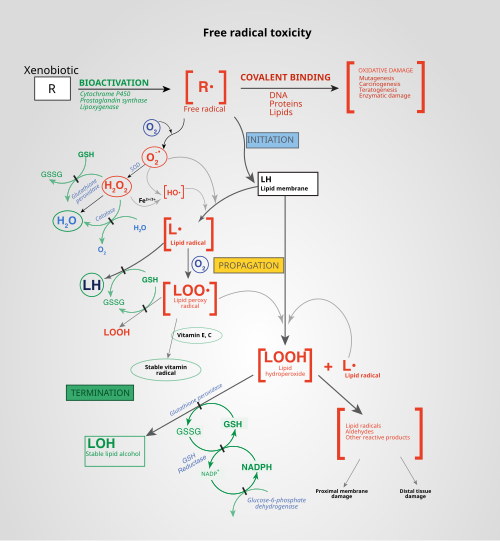
Figure 7 — ROS and Free Radical Toxicity. Mechanisms of free radical generation and the pathways by which reactive oxygen species damage cellular components through lipid peroxidation, protein oxidation, and DNA damage, along with the antioxidant defense systems that counteract them.
The Fenton reaction is the single most important mechanism for generating the highly destructive hydroxyl radical. This is why iron overload states (hemochromatosis, transfusional hemosiderosis) and copper excess (Wilson disease) cause tissue damage — free transition metals catalyze hydroxyl radical formation.
06 Necrosis: Types & Mechanisms
Necrosis is the morphologic pattern of cell death that occurs after irreversible injury in a living organism. It is characterized by enzymatic digestion of the cell (autolysis or heterolysis) and always elicits an inflammatory response due to leakage of cellular contents into the extracellular space.
Types of Necrosis
Type
Mechanism
Morphology
Classic Locations
Coagulative
Ischemia → protein denaturation preserves cell outlines; proteolytic enzymes denatured
Firm, pale tissue; ghost outlines of cells on H&E; preserved architecture
Diabetic foot; bowel ischemia; gas gangrene (Clostridium perfringens)
HIGH-YIELD DISTINCTION
Brain infarcts undergo liquefactive necrosis (not coagulative), making the brain the only solid organ exception to the “coagulative necrosis in solid organ infarcts” rule. The abundance of hydrolytic enzymes in neural tissue and the high lipid content favor enzymatic digestion.
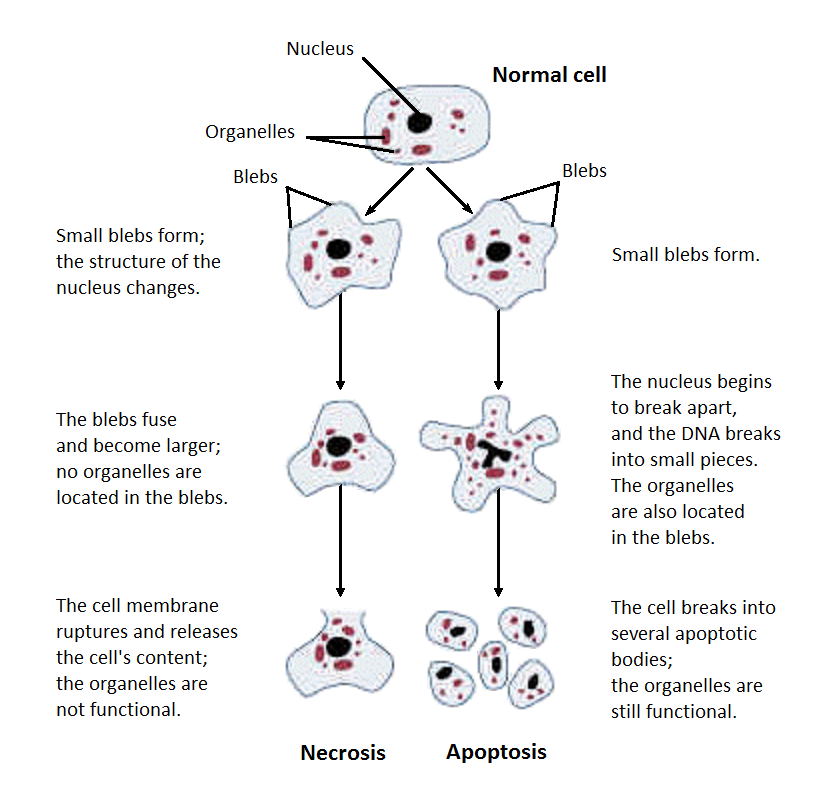
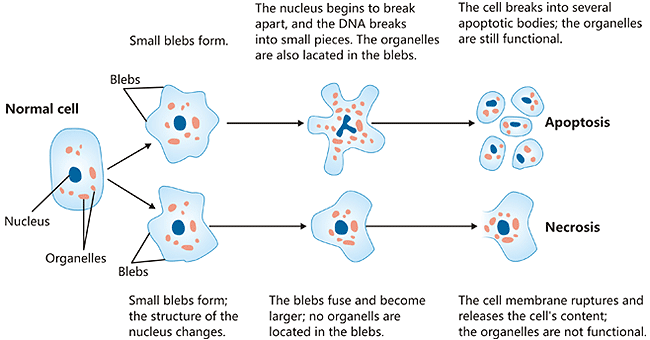
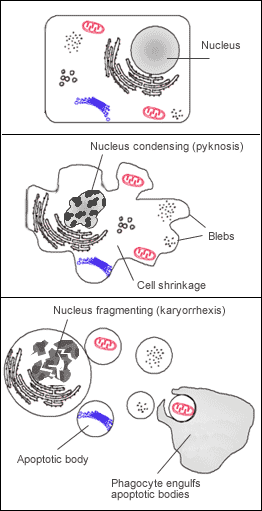
Figure 8 — Necrosis vs. Apoptosis. Structural alterations during necrosis compared to apoptosis. Necrosis features cell swelling, membrane disruption, and inflammatory response, while apoptosis involves cell shrinkage, chromatin condensation, and formation of apoptotic bodies without inflammation.
Nuclear Changes in Necrosis
Pyknosis — nuclear shrinkage and increased basophilia (chromatin condensation)
Karyorrhexis — fragmentation of the pyknotic nucleus
Karyolysis — dissolution of chromatin due to DNase activity; nucleus fades away
07 Apoptosis & Programmed Cell Death
Apoptosis is a tightly regulated, energy-dependent mechanism of programmed cell death that eliminates unwanted, damaged, or aged cells without eliciting an inflammatory response. Unlike necrosis, apoptotic cells shrink, their chromatin condenses, and they fragment into membrane-bound apoptotic bodies that are rapidly phagocytosed.
Apoptosis vs Necrosis
Feature
Apoptosis
Necrosis
Cell size
Shrinkage
Swelling (oncosis)
Membrane
Intact; phosphatidylserine flips to outer leaflet (“eat me” signal)
Disrupted; contents leak out
Nucleus
Fragmentation into nucleosomal-size fragments (DNA ladder on gel)
Pyknosis → karyorrhexis → karyolysis
Inflammation
Absent (no leakage of contents)
Present (DAMP release)
Energy
ATP-dependent (active process)
ATP-depleted (passive)
Mechanism
Caspase cascade
Enzymatic digestion by released lysosomal enzymes
Intrinsic (Mitochondrial) Pathway
Triggered by DNA damage, growth factor withdrawal, ER stress, or misfolded proteins. Pro-apoptotic BH3-only proteins (Bad, Bim, Bid) activate Bax and Bak, which oligomerize in the mitochondrial outer membrane, forming pores that release cytochrome c. Cytochrome c binds Apaf-1 to form the apoptosome, which activates caspase-9 (initiator caspase) → caspase-3 (executioner caspase). Anti-apoptotic proteins Bcl-2 and Bcl-xL prevent Bax/Bak oligomerization and are overexpressed in many cancers (e.g., follicular lymphoma with t(14;18)).
Figure 9 — Apoptosis Pathways. The intrinsic (mitochondrial) and extrinsic (death receptor) pathways of apoptosis. Both converge on executioner caspases (caspase-3, -6, -7) that dismantle the cell. The intrinsic pathway is regulated by the Bcl-2 family; the extrinsic pathway is initiated by death receptor signaling (Fas/FasL, TNF/TNFR).
Extrinsic (Death Receptor) Pathway
Initiated by binding of death ligands to death receptors: Fas ligand → Fas (CD95) or TNF → TNF receptor 1. Receptor trimerization recruits adaptor proteins (FADD) to form the death-inducing signaling complex (DISC), which activates caspase-8 (initiator) → caspase-3 (executioner). Both pathways converge on the executioner caspases (caspase-3, -6, -7) that cleave cytoskeletal proteins, nuclear lamins, and activate endonucleases (CAD/DFF40).
CLINICAL CORRELATION
Dysregulated apoptosis underlies many diseases: too little apoptosis → cancer (e.g., Bcl-2 overexpression in follicular lymphoma), autoimmune disease (failure to eliminate self-reactive lymphocytes); too much apoptosis → neurodegenerative diseases (Alzheimer, Parkinson), aplastic anemia, ischemia-reperfusion injury. The p53 tumor suppressor is a critical pro-apoptotic regulator — loss of p53 function (mutated in >50% of human cancers) impairs the cell’s ability to undergo apoptosis in response to DNA damage.
08 Intracellular Accumulations & Calcification
Abnormal intracellular accumulations result from excessive intake, abnormal metabolism, defective transport/secretion, or inability to degrade a substance. These accumulations provide important diagnostic clues on histopathology.
Types of Intracellular Accumulations
Substance
Mechanism
Example
Histologic Appearance
Lipid (steatosis)
Excess triglycerides in parenchymal cells (usually hepatocytes)
Alcoholic/non-alcoholic fatty liver disease
Clear vacuoles pushing nucleus to periphery (macrovesicular); or small droplets (microvesicular)
Iron storage in macrophages; local or systemic overload
Hemochromatosis; chronic hemorrhage; transfusions
Golden-brown granular pigment; Prussian blue stain positive
Lipofuscin
“Wear and tear” pigment from lipid peroxidation of membranes; not harmful
Aging (heart, liver, brain — “brown atrophy”)
Yellow-brown granular perinuclear pigment
Melanin
Endogenous pigment from melanocytes
Melanoma; nevi; melanosis coli
Brown-black pigment; bleached by melanin bleach
Carbon (anthracosis)
Inhaled carbon particles engulfed by macrophages
Coal workers’ pneumoconiosis; urban air pollution
Black pigment in lung macrophages and hilar lymph nodes
Figure 10 — Intracellular Accumulations. Histologic examples of abnormal intracellular accumulations including lipid droplets, protein inclusions, and pigment deposits that provide important diagnostic clues in pathology specimens.
Pathologic Calcification
Type
Serum Ca2+
Mechanism
Examples
Dystrophic
Normal
Calcium deposits in dead/dying tissue; nucleation on membrane-bound phospholipids or denatured proteins
Calcium deposits in normal tissue due to systemic hypercalcemia
Hyperparathyroidism; renal failure (secondary hyperPTH); sarcoidosis; metastatic bone destruction; vitamin D excess
Psammoma bodies are concentric, laminated calcifications found in papillary thyroid carcinoma, papillary serous cystadenocarcinoma of the ovary, meningioma, and mesothelioma. Their presence on cytology or biopsy is a useful diagnostic clue (“PSaMMoma” mnemonic: Papillary thyroid, Serous ovarian, Meningioma, Mesothelioma).
09 Acute Inflammation
Acute inflammation is the rapid, initial response to tissue injury or infection, characterized by vasodilation, increased vascular permeability, and recruitment of leukocytes (predominantly neutrophils). It occurs within minutes to hours and typically resolves within days.
Cardinal Signs of Inflammation (Celsus + Virchow)
Sign
Latin
Mechanism
Redness
Rubor
Vasodilation → increased blood flow
Heat
Calor
Vasodilation → warm blood to surface
Swelling
Tumor
Increased vascular permeability → exudate
Pain
Dolor
Bradykinin, PGE2 sensitize nociceptors; pressure from edema
Loss of function
Functio laesa
Pain, swelling, tissue destruction impair function
Increased vascular permeability — endothelial cell contraction creates inter-endothelial gaps in post-capillary venules; allows protein-rich exudate to enter interstitium
Stasis — fluid loss concentrates RBCs, increasing viscosity and slowing flow; facilitates leukocyte margination
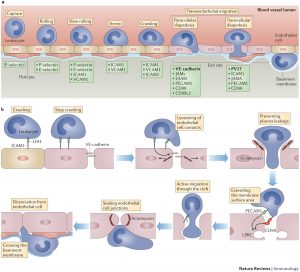
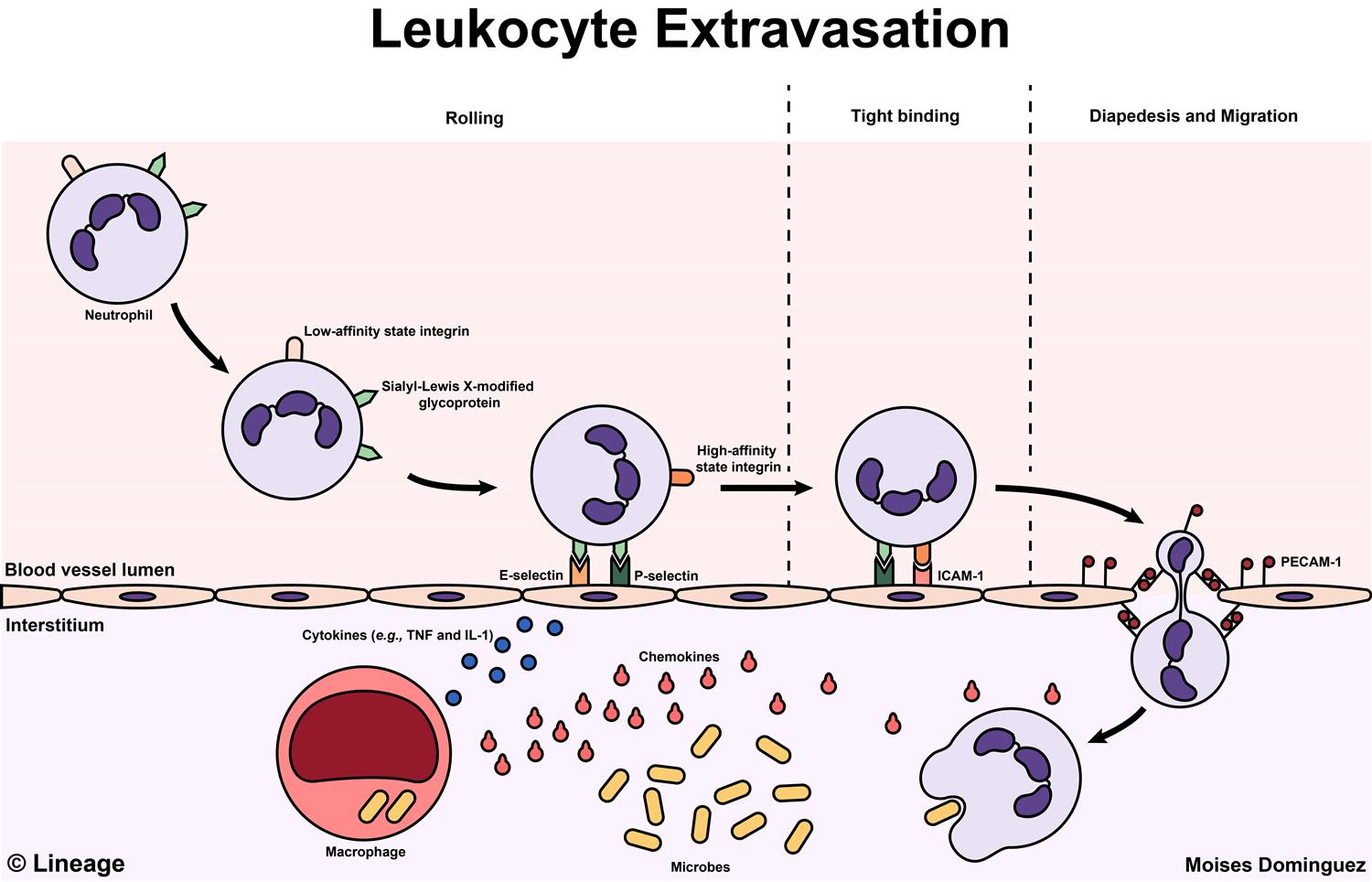
Figure 11 — Leukocyte Extravasation Overview. Diagram illustrating the molecules involved in leukocyte extravasation from the vascular lumen into tissues, including cadherins, integrins (LFA-1, Mac-1), selectins, and their endothelial ligands (ICAM-1, VCAM-1, PECAM-1).
Leukocyte Recruitment Cascade
STEPS OF LEUKOCYTE EXTRAVASATION
Margination — slowed blood flow allows WBCs to move to vessel periphery
Rolling — loose, transient adhesion via selectins (E-selectin, P-selectin on endothelium; L-selectin on leukocytes) binding sialyl-Lewis X carbohydrates
Firm adhesion — chemokine-activated integrins (LFA-1/Mac-1) on leukocytes bind ICAM-1 and VCAM-1 on endothelium
Transmigration (diapedesis) — leukocytes squeeze between endothelial cells via PECAM-1 (CD31) interactions, traversing the basement membrane with collagenases
Chemotaxis — directed migration along chemical gradient (C5a, LTB4, IL-8, bacterial peptides [fMLP])
Figure 12 — Steps of Leukocyte Extravasation. The sequential steps of leukocyte recruitment: margination, selectin-mediated rolling, chemokine-activated integrin-mediated firm adhesion, and PECAM-1-mediated transmigration (diapedesis) through the endothelium into inflamed tissue.
Leukocyte adhesion deficiency type 1 (LAD-1) is caused by a defect in the CD18 β2-integrin subunit, preventing firm adhesion. Patients have markedly elevated WBC counts (neutrophilia — cells cannot leave the bloodstream), recurrent severe bacterial infections, impaired wound healing, and delayed umbilical cord separation.
Phagocytosis & Killing
Neutrophils and macrophages recognize pathogens via opsonin receptors (Fc receptor for IgG, C3b receptor/CR1), pattern recognition receptors (TLRs for PAMPs), and mannose receptors. Engulfment creates a phagosome that fuses with lysosomes. Killing mechanisms include:
Oxygen-independent: lysozyme, lactoferrin, defensins, major basic protein (eosinophils), bactericidal/permeability-increasing protein (BPI)
Chronic granulomatous disease (CGD) results from defective NADPH oxidase (most commonly X-linked defect in gp91phox). Patients cannot generate the respiratory burst and are susceptible to catalase-positive organisms (S. aureus, Aspergillus, Serratia, Nocardia, Burkholderia cepacia) because catalase-positive organisms destroy their own H2O2, removing the substrate that could otherwise fuel the MPO system. Catalase-negative organisms (Streptococci) produce H2O2 that CGD neutrophils can still use. Diagnosed by dihydrorhodamine (DHR) flow cytometry or nitroblue tetrazolium (NBT) test.
10 Chemical Mediators of Inflammation
Inflammatory mediators are derived from plasma proteins or cells and orchestrate every step of the inflammatory response. They are produced in response to tissue injury, PAMPs, and DAMPs, and their actions are tightly regulated to limit collateral tissue damage.
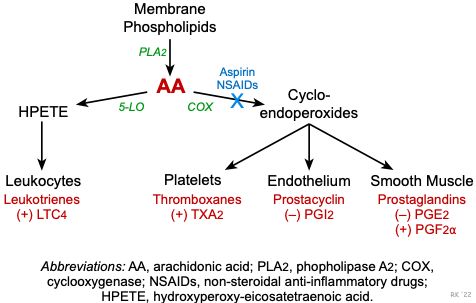
Figure 13 — Arachidonic Acid Metabolism Pathway. The COX and lipoxygenase (LOX) pathways of arachidonic acid metabolism. Phospholipase A2 liberates arachidonic acid from membrane phospholipids; COX produces prostaglandins and thromboxane, while 5-LOX produces leukotrienes. Key pharmacologic targets are indicated.
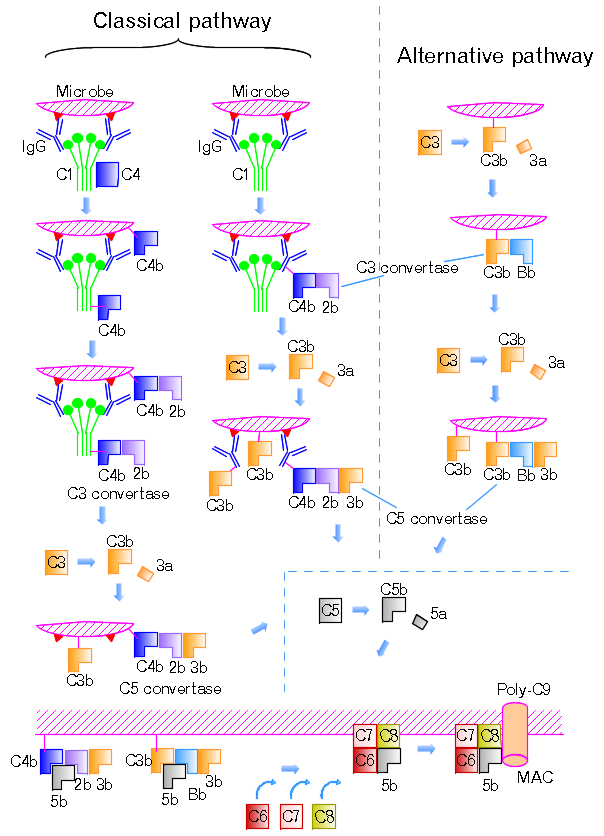
Plasma-Derived Mediators: Complement System
The complement system comprises >30 plasma proteins activated by three pathways: classical (antibody-antigen complexes → C1q binding), alternative (spontaneous C3 hydrolysis on microbial surfaces; no antibody needed), and lectin (mannose-binding lectin binds microbial mannose residues). All three pathways converge at C3 convertase, which cleaves C3 into C3a and C3b — the central event in complement activation.
Component
Function
Clinical Deficiency
C3a, C5a
Anaphylatoxins — mast cell degranulation, vasodilation, increased permeability; C5a is also the most potent neutrophil chemotactic factor
C3 deficiency: severe recurrent pyogenic infections + immune complex disease (C3 is the convergence point)
C3b
Opsonization — facilitates phagocytosis by macrophages and neutrophils
C5b-9 (MAC)
Membrane attack complex — lysis of target cells by forming transmembrane pores
C5–C9 deficiency: increased susceptibility to Neisseria infections (meningococcal meningitis/sepsis)
C1 esterase inhibitor
Regulates classical pathway and kinin system
Hereditary angioedema (HAE): recurrent episodes of non-pruritic, non-pitting edema of face, larynx, and bowel; does NOT respond to epinephrine or antihistamines
C1q, C2, C4
Early classical pathway components
C2 deficiency (most common complement deficiency): SLE-like illness due to impaired immune complex clearance
DAF (CD55), MIRL (CD59)
GPI-anchored complement regulatory proteins on cell surfaces; prevent MAC assembly on self cells
Paroxysmal nocturnal hemoglobinuria (PNH): loss of GPI anchor (PIGA mutation) → complement-mediated hemolysis, thrombosis, pancytopenia
The most common complement deficiency is C2 deficiency, which presents with SLE-like illness. However, the most clinically devastating is C3 deficiency (C3 is the convergence point of all pathways), and the most testable association is terminal complement (C5–C9) deficiency with recurrent Neisseria infections. Any patient with recurrent meningococcal infections should be evaluated for complement deficiency.
Figure 14 — Complement Pathways. The three activation pathways of the complement system: classical (C1q-antibody complex), alternative (spontaneous C3 hydrolysis), and lectin (mannose-binding lectin). All converge at C3 convertase, generating C3a/C5a (anaphylatoxins), C3b (opsonin), and the membrane attack complex (C5b-9).
Kinin System
Factor XII (Hageman factor) activates prekallikrein → kallikrein, which cleaves high-molecular-weight kininogen to produce bradykinin. Bradykinin causes vasodilation, increased vascular permeability, pain, and bronchoconstriction. It is inactivated by ACE (kininase II) — this is why ACE inhibitors can cause angioedema and dry cough (via accumulated bradykinin).
Chronic inflammation is a prolonged inflammatory response (weeks to years) characterized by simultaneous tissue destruction and repair. The dominant cell types shift from neutrophils to macrophages, lymphocytes, and plasma cells. It may follow acute inflammation or arise de novo.
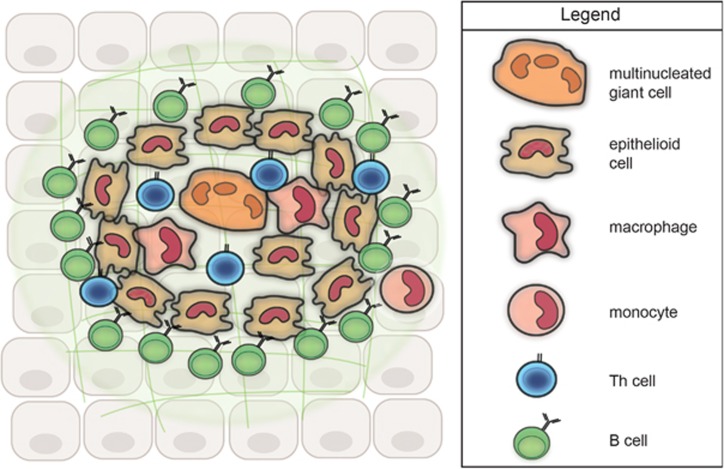
A distinctive pattern of chronic inflammation characterized by aggregates of activated macrophages that transform into epithelioid cells (elongated macrophages with pale pink cytoplasm), often with multinucleated giant cells (Langhans type with peripheral horseshoe nuclei, or foreign-body type with scattered nuclei). Granulomas may be caseating (central necrosis, classic for TB) or non-caseating (sarcoidosis, Crohn disease, berylliosis).
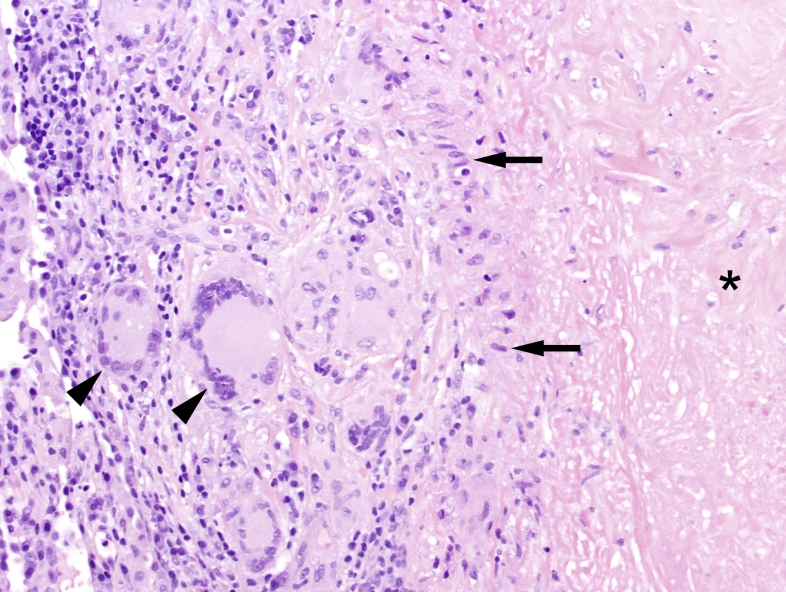
Figure 15 — Granuloma Cellular Organization. Model of granuloma structure showing the organized aggregate of epithelioid macrophages, multinucleated giant cells, and a peripheral cuff of lymphocytes. T helper 1 cells secrete IFN-gamma to activate macrophages, and TNF-alpha is critical for granuloma maintenance.Figure 16 — Caseating Granuloma (Tuberculosis). Histopathology of a necrotizing (caseating) granuloma in tuberculosis showing central caseous necrosis surrounded by epithelioid histiocytes and Langhans-type giant cells with a peripheral lymphocytic cuff.Figure 17 — Non-Caseating Granulomas (Sarcoidosis). Non-necrotizing granulomas characteristic of sarcoidosis, composed of tightly clustered epithelioid histiocytes without central necrosis. Sarcoidosis is the most common cause of non-caseating granulomas.
Causes of Granulomatous Inflammation
Caseating Granulomas
Non-Caseating Granulomas
Tuberculosis (most common worldwide)
Sarcoidosis (most common cause of non-caseating granulomas)
Wegener granulomatosis (granulomatosis with polyangiitis)
The TH1 immune response drives granuloma formation: macrophages present antigen → TH1 cells secrete IFN-γ → macrophages activated to epithelioid cells. TNF-α is critical for maintaining granuloma integrity. This is why anti-TNF therapy (infliximab, adalimumab) requires TB screening before initiation — blocking TNF can reactivate latent TB by disrupting granuloma containment.
12 Tissue Repair, Regeneration & Fibrosis
After inflammation, tissue integrity is restored by regeneration (replacement of damaged cells with cells of the same type) or repair by connective tissue (scar formation/fibrosis). The outcome depends on the tissue’s regenerative capacity and the extent of damage.
Cell Proliferative Capacity
Category
Definition
Examples
Labile cells
Continuously dividing throughout life
Skin epidermis, GI epithelium, hematopoietic cells, cervical epithelium
Inflammation (1–3 days): Neutrophils clear debris (peak day 1–2); macrophages arrive (peak day 3–5) — macrophages are the most important cell in wound healing
Proliferation (3–21 days): Granulation tissue forms (new capillaries via angiogenesis + fibroblasts producing collagen type III); epithelial migration covers wound surface
Remodeling (weeks to months): Type III collagen replaced by type I collagen; wound strength increases to maximum ~80% of original (never reaches 100%)
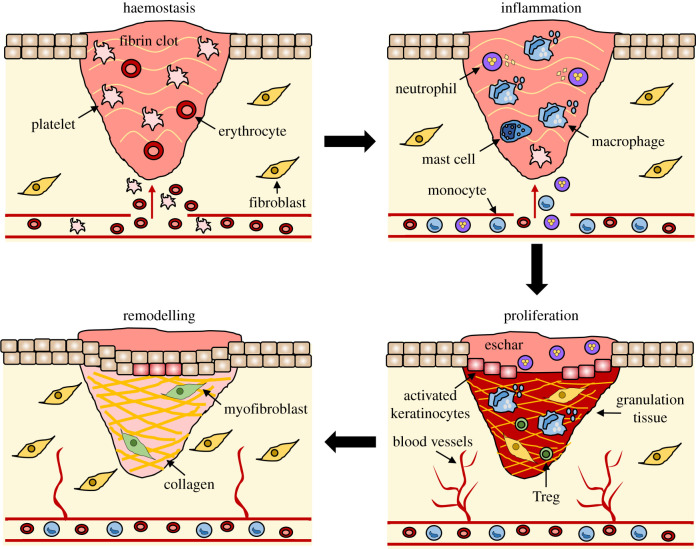
Figure 18 — Stages of Wound Repair. The four overlapping phases of wound healing: hemostasis (platelet plug, fibrin clot), inflammation (neutrophil and macrophage recruitment), proliferation (granulation tissue, angiogenesis, re-epithelialization), and remodeling (collagen maturation, scar formation).
Growth Factors in Repair
Factor
Source
Action
PDGF
Platelets, macrophages
Fibroblast and smooth muscle chemotaxis and proliferation
TGF-β
Platelets, macrophages, T cells
Fibroblast chemotaxis; stimulates collagen synthesis; anti-inflammatory; key driver of fibrosis
VEGF
Macrophages, keratinocytes
Angiogenesis (new blood vessel formation)
FGF
Macrophages, fibroblasts
Angiogenesis; fibroblast proliferation
EGF
Platelets, macrophages, saliva
Epithelial and fibroblast proliferation
Factors That Impair Wound Healing
Factor
Mechanism of Impairment
Infection
Most important local cause; persistent inflammation delays repair; increases tissue destruction
Persistent inflammation and granuloma formation; nidus for infection
Ischemia / Poor perfusion
Peripheral vascular disease, venous stasis; oxygen is essential for collagen hydroxylation and leukocyte killing
Zinc deficiency
Zinc is a cofactor for collagenase and metalloproteinases needed in remodeling
Copper deficiency
Copper is cofactor for lysyl oxidase, which cross-links collagen
Figure 19 — Wound Healing Regulators. Key molecular, biological, and mechanical factors that modulate each phase of wound healing. Dysregulation of these factors can lead to either impaired healing (chronic wounds) or excessive scarring (keloids, hypertrophic scars, fibrosis).
Abnormal Wound Healing
Keloid — excessive collagen deposition that extends beyond the original wound borders; more common in African Americans; does not regress spontaneously; recurs after excision; type III and type I collagen
Hypertrophic scar — excessive collagen but remains within wound borders; may regress over time
Dehiscence — wound rupture, most common at abdominal surgical sites; risk factors: obesity, increased abdominal pressure, infection, poor nutrition
Contracture — exaggerated wound contraction causing deformity; common after burns; myofibroblasts are responsible
The tensile strength of a wound reaches only about 80% of normal even after complete healing and remodeling, which is why surgical incisions and healed wounds remain vulnerable to re-injury. Collagen cross-linking is the primary determinant of tensile strength, requiring vitamin C (for prolyl and lysyl hydroxylase) and copper (for lysyl oxidase).
13 Edema & Fluid Dynamics
Edema is the accumulation of excess fluid in the interstitial space (or body cavities, where it is termed effusion). Understanding the Starling forces is essential to comprehending the pathophysiology of edema formation.
Starling Forces
Force
Normal Value (capillary end)
Effect
Capillary hydrostatic pressure (Pc)
~35 mmHg (arterial), ~15 mmHg (venous)
Pushes fluid OUT of capillary
Interstitial hydrostatic pressure (Pi)
~0 mmHg
Pushes fluid INTO capillary (opposing Pc)
Plasma oncotic pressure (πc)
~25 mmHg
Pulls fluid INTO capillary (albumin-dependent)
Interstitial oncotic pressure (πi)
~1 mmHg
Pulls fluid OUT of capillary
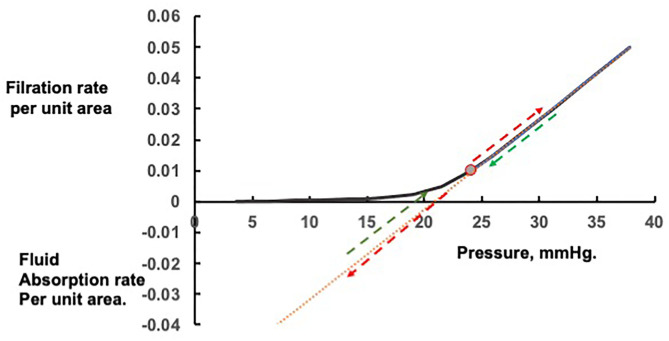
Figure 20 — Starling Forces and Capillary Fluid Exchange. The balance of hydrostatic and oncotic pressures that governs fluid movement across capillary walls. Disruption of these Starling forces through increased hydrostatic pressure, decreased oncotic pressure, or increased permeability leads to edema formation.
Pathophysiologic Mechanisms of Edema
Mechanism
Pathophysiology
Examples
Increased hydrostatic pressure
Elevated venous pressure transmits retrograde to capillary bed
Dependent edema in CHF is pitting edema (finger pressure leaves a temporary depression). Lymphedema is characteristically non-pitting because the interstitial fluid is protein-rich and undergoes fibrosis over time. This distinction helps identify the underlying mechanism at the bedside.
SAAG = serum albumin − ascites albumin. SAAG ≥1.1 g/dL indicates portal hypertension (cirrhosis, CHF, Budd-Chiari) with 97% accuracy. SAAG <1.1 g/dL indicates non-portal hypertensive causes (peritoneal carcinomatosis, TB peritonitis, nephrotic syndrome, pancreatitis). SAAG has replaced the older transudate/exudate classification for ascitic fluid.
14 Thrombosis & Virchow’s Triad
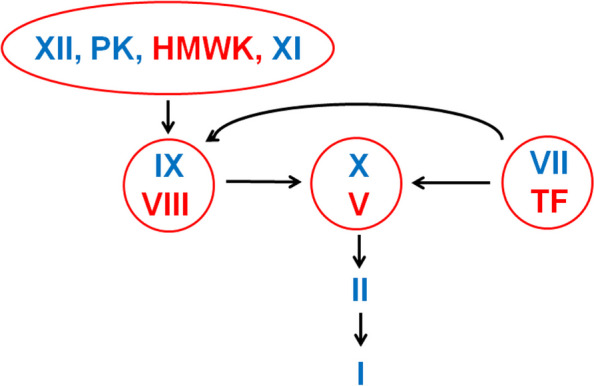
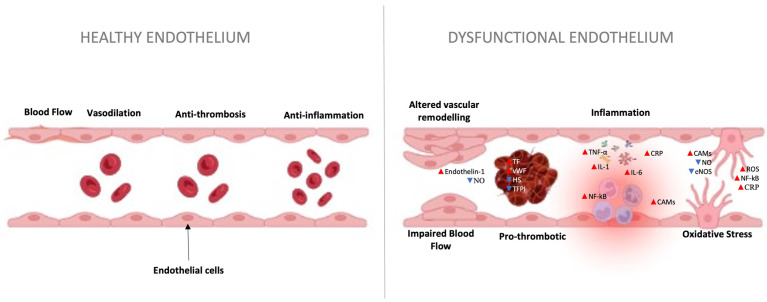
Thrombosis is the pathologic formation of a blood clot (thrombus) within an intact blood vessel. It is governed by Virchow’s triad: (1) endothelial injury, (2) stasis or turbulence, and (3) hypercoagulability.
Figure 21 — Virchow’s Triad. The three factors predisposing to thrombosis: endothelial injury (the most important factor), stasis or turbulent blood flow, and hypercoagulability. Clinical thrombosis typically involves at least two of these three elements.
Virchow’s Triad
Factor
Mechanism
Clinical Examples
Endothelial injury
Loss of anti-thrombotic properties; exposure of subendothelial collagen and tissue factor
Factor V Leiden; prothrombin 20210A; antithrombin III deficiency; protein C/S deficiency; antiphospholipid syndrome; malignancy (Trousseau syndrome); OCPs; nephrotic syndrome
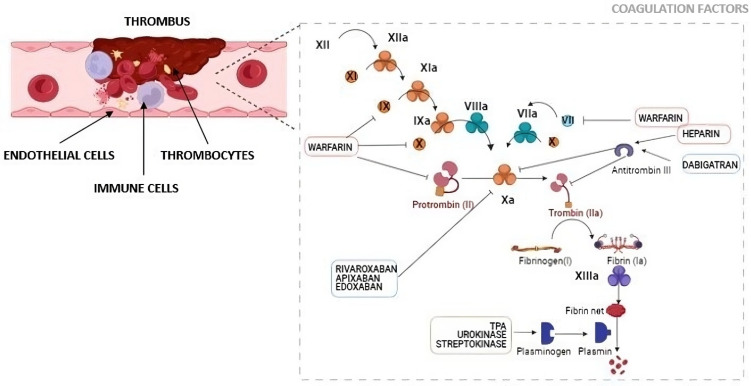
Figure 22 — Coagulation Cascade. The coagulation pathway from tissue factor initiation through thrombin generation to fibrin clot formation. The intrinsic and extrinsic pathways converge at factor X activation, leading to the common pathway and ultimately cross-linked fibrin.
Hereditary Thrombophilias
Disorder
Defect
Inheritance
Key Points
Factor V Leiden
Factor V resistant to inactivation by activated protein C
AD
Most common hereditary thrombophilia (5% of Caucasians); heterozygous = 5× risk; homozygous = 50× risk of VTE
Heparin resistance (heparin requires ATIII to work)
Protein C deficiency
Cannot inactivate factors Va and VIIIa
AD
Warfarin-induced skin necrosis (protein C has short half-life, drops first)
Protein S deficiency
Protein S is cofactor for protein C
AD
Similar phenotype to protein C deficiency
Warfarin skin necrosis occurs during the first few days of warfarin initiation because protein C (an anticoagulant with a short half-life of ~8 hours) is depleted faster than the procoagulant factors (factor II t1/2 = 60 hours), creating a transient hypercoagulable state. This is why heparin bridging is essential when starting warfarin, especially in patients with known protein C or S deficiency.
15 Embolism
An embolus is a detached intravascular mass (solid, liquid, or gaseous) carried by the blood to a site distant from its point of origin, where it lodges and obstructs a vessel. Approximately 99% of emboli arise from thrombi (thromboembolism).
Figure 23 — Thromboembolism Pathophysiology. Coagulation and clotting factor signaling in thrombus formation. Over 95% of pulmonary emboli originate from deep vein thrombi in the lower extremities, traveling through the IVC and right heart to lodge in the pulmonary arteries.
Types of Embolism
Type
Source / Mechanism
Consequences
Pulmonary thromboembolism (PE)
>95% from deep veins of legs (DVT); travels through IVC → right heart → pulmonary arteries
Saddle embolus → sudden death (obstructs bifurcation); medium arteries → pulmonary infarction (wedge-shaped, hemorrhagic); small arteries → may be silent or cause pulmonary HTN if recurrent
Systemic (arterial) thromboembolism
~80% from intracardiac mural thrombi (LV wall post-MI, LA in atrial fibrillation); also aortic aneurysms, atherosclerotic plaque
Surgery, trauma, IV access, decompression sickness (“the bends”)
>100 mL needed to cause symptoms; air locks in right ventricle
Amniotic fluid embolism
Amniotic fluid enters maternal circulation during labor/delivery or C-section
Sudden dyspnea, shock, DIC, seizures; 80% mortality; squamous cells and fetal debris in pulmonary vessels
Cholesterol / atheroemboli
Cholesterol crystals dislodge from ulcerated atherosclerotic plaques
“Blue toe syndrome”; livedo reticularis; renal failure; biconvex cleft-shaped spaces on biopsy
Figure 24 — Endothelial Effects of Pulmonary Embolism. The pathophysiologic effects of pulmonary embolism on the vascular endothelium, including disruption of normal endothelial function, platelet activation, inflammatory cascade initiation, and right ventricular pressure overload.
HIGH-YIELD: PARADOXICAL EMBOLISM
A venous thrombus can reach the systemic arterial circulation through a right-to-left shunt, most commonly a patent foramen ovale (PFO), which is present in ~25% of adults. This is called a paradoxical embolism and should be suspected in a young patient with a cryptogenic stroke and DVT. Diagnosis: transesophageal echocardiography with agitated saline (“bubble study”) showing early bubble transit from RA to LA.
16 Infarction & Ischemia
An infarct is an area of ischemic necrosis caused by occlusion of the arterial supply or (less commonly) venous drainage. Infarcts are classified as white (anemic/pale) or red (hemorrhagic) based on the amount of hemorrhage and the tissue architecture.
White vs Red Infarcts
Feature
White (Anemic) Infarct
Red (Hemorrhagic) Infarct
Mechanism
Arterial occlusion in solid organs with single (end-artery) blood supply
Venous occlusion; arterial occlusion in loose tissue with dual blood supply; reperfusion of previously ischemic tissue
Pale, wedge-shaped (base at capsule, apex at occlusion site)
Dark red, hemorrhagic, irregular borders
Ischemia-Reperfusion Injury
Paradoxically, restoration of blood flow after ischemia can exacerbate tissue damage beyond what occurred during the ischemic period. Mechanisms include:
ROS burst — re-oxygenation generates massive free radicals from damaged mitochondria, xanthine oxidase, and recruited neutrophils
Calcium overload — reperfusion floods cells with Ca2+
Neutrophil influx — restored flow delivers activated neutrophils that release proteases and ROS
Ischemia-reperfusion injury is clinically significant in myocardial infarction (post-PCI reperfusion arrhythmias), organ transplantation (cold ischemia time), and stroke (hemorrhagic transformation after thrombolysis). In cardiac surgery, cardioplegia solutions are designed to minimize this injury.
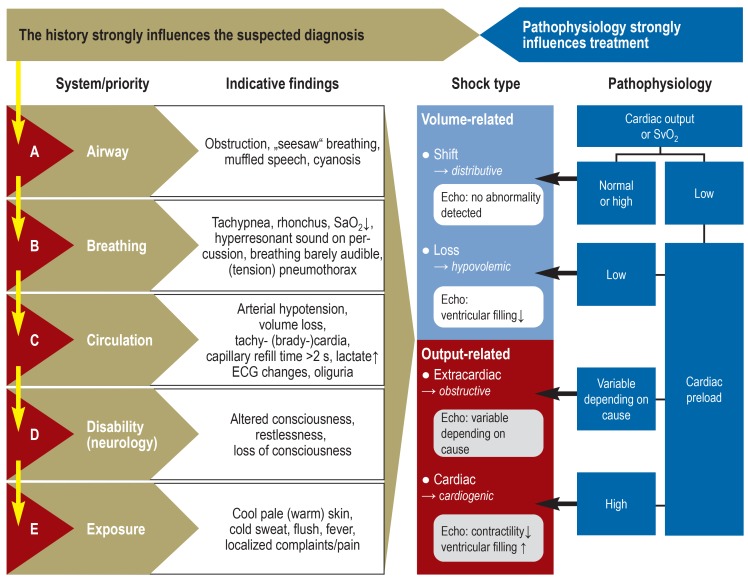
17 Shock & Hemodynamic Collapse
Shock is a state of systemic hypoperfusion due to reduced cardiac output or reduced effective circulating blood volume, resulting in inadequate tissue oxygenation and cellular hypoxia. If uncorrected, shock progresses to irreversible organ damage and death.
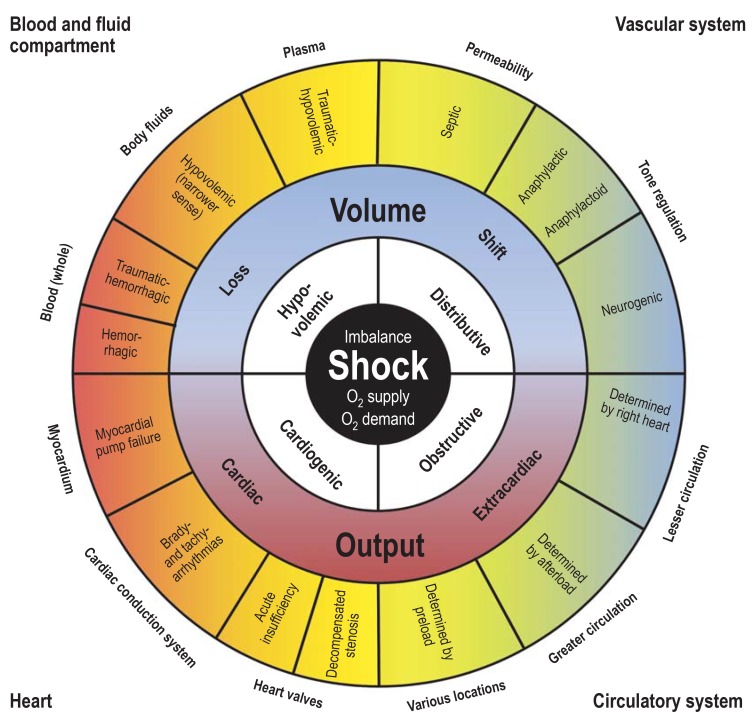
Figure 25 — Classification of Shock. The four major categories of shock: cardiogenic (pump failure), hypovolemic (volume loss), distributive (vasodilation, as in sepsis and anaphylaxis), and obstructive (mechanical obstruction to flow). Each type has distinct hemodynamic profiles.
Types of Shock
Type
Mechanism
CO
SVR
PCWP
Examples
Cardiogenic
Pump failure — heart cannot generate adequate CO
↓
↑
↑
Massive MI (>40% LV); acute mitral regurgitation; cardiac tamponade; myocarditis
Hypovolemic
Decreased blood or plasma volume
↓
↑
↓
Hemorrhage (trauma, GI bleed); burns; severe dehydration; third-spacing
Distributive (septic)
Systemic vasodilation; maldistribution of blood flow
↑ (early, “warm shock”)
↓↓
↓ or N
Sepsis (most common cause of death in ICU); anaphylaxis; neurogenic shock (spinal cord injury)
Compensated (non-progressive): Neurohumoral reflexes maintain perfusion — tachycardia, vasoconstriction, RAAS activation, ADH release; BP may be near normal; reversible
Figure 26 — Shock Diagnostic Algorithm. Clinical algorithm for the differential diagnosis of shock based on hemodynamic parameters, physical examination findings, and laboratory data to distinguish cardiogenic, hypovolemic, distributive, and obstructive etiologies.
Septic Shock Pathophysiology
Bacterial products (LPS/endotoxin from gram-negatives, lipoteichoic acid from gram-positives) activate innate immune cells via TLR4/TLR2 → massive cytokine release (“cytokine storm”: TNF-α, IL-1, IL-6). This produces: (1) systemic vasodilation (NO-mediated) → hypotension; (2) endothelial activation → increased permeability, edema, DIC; (3) myocardial depression; (4) metabolic derangements (insulin resistance, hyperglycemia). Early septic shock is “warm shock” with vasodilation and high CO; late septic shock transitions to “cold shock” with myocardial depression and low CO.
Anaphylactic Shock
A subset of distributive shock caused by systemic type I hypersensitivity (IgE-mediated mast cell degranulation). Massive histamine and leukotriene release causes: profound vasodilation → hypotension; bronchoconstriction → respiratory distress; laryngeal edema → airway obstruction; urticaria and angioedema. Treatment: intramuscular epinephrine (0.3–0.5 mg IM in anterolateral thigh) is the first-line and most important intervention. Epinephrine reverses vasodilation (α-1), bronchoconstriction (β-2), and stabilizes mast cells (β-2). Secondary agents: IV fluids, H1/H2 blockers, corticosteroids (prevent late-phase reaction), and albuterol for persistent bronchospasm.
Neurogenic Shock
Caused by disruption of sympathetic outflow, typically from spinal cord injury above T6. Loss of sympathetic tone produces vasodilation (decreased SVR) and bradycardia (unopposed vagal tone). Unlike other forms of shock, neurogenic shock presents with warm, dry skin and bradycardia (rather than cool, clammy skin and tachycardia). Treatment: IV fluids + vasopressors (norepinephrine or phenylephrine) + atropine for significant bradycardia.
The Surviving Sepsis Campaign emphasizes the Hour-1 Bundle: measure lactate, obtain blood cultures before antibiotics, administer broad-spectrum antibiotics, begin rapid fluid resuscitation with 30 mL/kg crystalloid for hypotension or lactate ≥4, and apply vasopressors (norepinephrine first-line) if hypotension persists after fluids. Each hour of delay in antibiotics increases mortality.
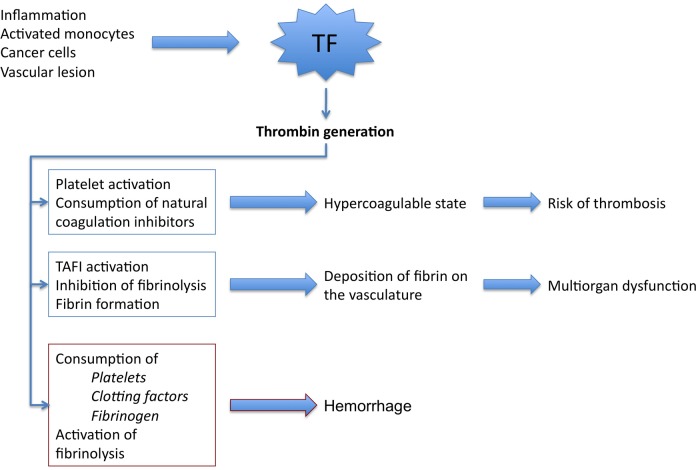
18 Disseminated Intravascular Coagulation (DIC)
DIC is a consumptive coagulopathy characterized by widespread activation of the coagulation cascade, leading to formation of microthrombi throughout the vasculature with simultaneous consumption of platelets and clotting factors, resulting paradoxically in both thrombosis and hemorrhage.
Pathophysiology
Triggering event releases tissue factor or other procoagulants into the circulation → widespread thrombin generation → fibrin deposition in microvasculature → consumption of platelets, fibrinogen, and clotting factors (II, V, VIII) → secondary fibrinolysis (plasmin activation) generates fibrin degradation products (FDPs) including D-dimers, which further impair platelet function and fibrin polymerization.
Figure 27 — DIC Pathogenesis. Pathogenetic pathways in disseminated intravascular coagulation. Tissue factor overexpression leads to explosive thrombin generation, widespread fibrin deposition in the microvasculature, consumption of clotting factors and platelets, and secondary fibrinolysis with D-dimer elevation.
Causes of DIC
Category
Examples
Obstetric
Placental abruption; amniotic fluid embolism; eclampsia; retained dead fetus
Mechanical shearing on fibrin strands in microvasculature (microangiopathic hemolytic anemia)
Thrombin time
↑
Low fibrinogen + FDP interference
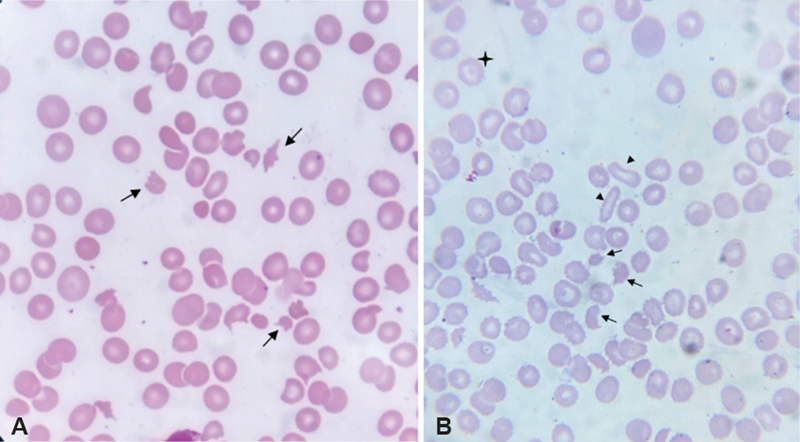
Figure 28 — Schistocytes on Peripheral Blood Smear. Peripheral blood smear showing schistocytes (fragmented red blood cells, arrows) characteristic of microangiopathic hemolytic anemia seen in DIC, TTP, and HUS. The mechanical shearing of RBCs on fibrin strands within the microvasculature produces these diagnostic fragments.
The combination of schistocytes on blood smear + thrombocytopenia + elevated D-dimer + prolonged PT/aPTT + low fibrinogen is virtually diagnostic of DIC. In acute promyelocytic leukemia (APL), DIC is the most common cause of early death, and treatment with all-trans retinoic acid (ATRA) is initiated immediately upon suspicion of APL even before confirmatory testing, because it induces differentiation and rapidly improves the coagulopathy.
19 Neoplasia Fundamentals & Nomenclature
Neoplasia (“new growth”) refers to unregulated cell proliferation that is autonomous and persists after removal of the inciting stimulus. A neoplasm (tumor) consists of neoplastic cells and supportive stroma (connective tissue and blood vessels).
Benign vs Malignant Neoplasms
Feature
Benign
Malignant
Differentiation
Well-differentiated; resembles tissue of origin
Variable; ranges from well-differentiated to anaplastic
Growth rate
Usually slow
Variable; often rapid
Growth pattern
Expansile, often encapsulated
Infiltrative, invasive, often not encapsulated
Metastasis
Absent
Present (defines malignancy)
Mitotic rate
Low
Often high; atypical mitoses
Nuclear features
Normal N:C ratio
Pleomorphism, hyperchromasia, high N:C ratio
Tumor Nomenclature
Tissue of Origin
Benign
Malignant
Epithelial — glandular
Adenoma
Adenocarcinoma
Epithelial — squamous
Squamous papilloma
Squamous cell carcinoma
Mesenchymal — bone
Osteoma
Osteosarcoma
Mesenchymal — cartilage
Chondroma
Chondrosarcoma
Mesenchymal — fat
Lipoma
Liposarcoma
Mesenchymal — smooth muscle
Leiomyoma
Leiomyosarcoma
Mesenchymal — skeletal muscle
Rhabdomyoma
Rhabdomyosarcoma
Mesenchymal — blood vessels
Hemangioma
Angiosarcoma
Lymphoid
—
Lymphoma / Leukemia
Melanocytes
Nevus
Melanoma
Germ cells
Mature teratoma
Immature teratoma; seminoma; choriocarcinoma
NAMING EXCEPTIONS (“-OMA” BUT MALIGNANT)
Several malignant tumors retain the “-oma” suffix despite being malignant: lymphoma, melanoma, mesothelioma, seminoma, hepatoblastoma, glioblastoma. These are important board-tested exceptions to the standard nomenclature rules.
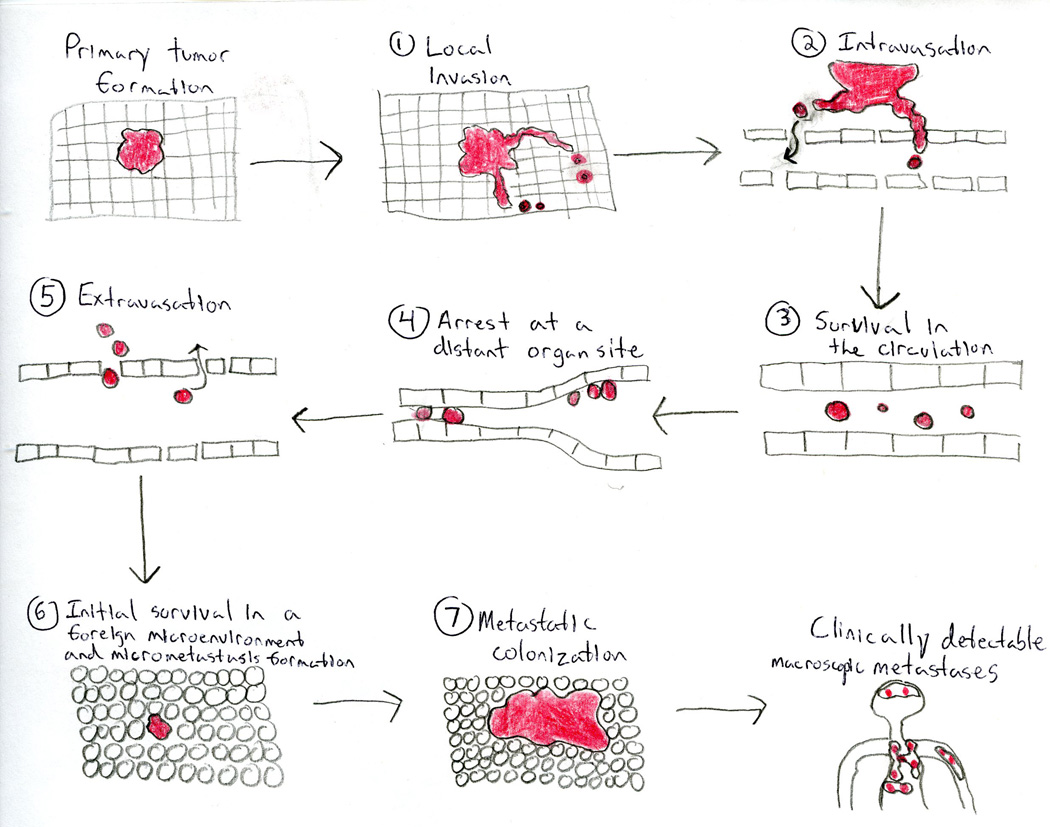
Routes of Metastasis
Route
Mechanism
Classic Examples
Lymphatic spread
Most common initial route for carcinomas; tumor cells invade lymphatic channels and colonize regional lymph nodes
Breast CA → axillary nodes; lung CA → mediastinal nodes; colorectal CA → mesenteric nodes; Virchow node (left supraclavicular) = gastric CA
Hematogenous spread
Most common route for sarcomas; tumor cells enter bloodstream; venous drainage determines metastatic site
Renal cell CA → IVC → lung; colorectal CA → portal vein → liver; prostate CA → Batson vertebral venous plexus → vertebral mets
Seeding of body cavities
Direct spread across serosal surfaces
Ovarian CA → peritoneal carcinomatosis (omental caking); lung CA → malignant pleural effusion
Figure 29 — The Invasion-Metastasis Cascade. Steps of tumor metastasis: local invasion through basement membrane and stroma, intravasation into blood or lymphatic vessels, survival in the circulation, arrest at distant sites, extravasation, and colonization to form secondary tumors.
Common Sites of Metastasis by Primary Cancer
Primary Cancer
Most Common Metastatic Sites
Lung
Brain, bone, liver, adrenal glands (most common cancer to metastasize to adrenal)
Breast
Bone (most common), lung, liver, brain
Colon
Liver (via portal circulation; most common cancer to metastasize to liver), lung
Prostate
Bone (osteoblastic/sclerotic mets via Batson plexus)
Renal cell
Lung, bone, brain (can invade renal vein and IVC)
Melanoma
Can metastasize to virtually any organ; brain mets very common
Carcinomas spread first by lymphatics (sentinel node biopsy concept); sarcomas spread first by blood (hematogenous). The most common overall site of distant metastasis is the liver (portal drainage from GI tract), followed by lung. The most common primary malignancy of bone in adults is metastatic disease (not primary bone tumors), with breast, prostate, lung, kidney, and thyroid being the most common sources.
20 Hallmarks of Cancer & Molecular Oncology
The Hallmarks of Cancer (Hanahan & Weinberg, 2000; updated 2011) define the fundamental capabilities acquired during multistep tumorigenesis.
Cell cycle arrest (G1/S checkpoint); DNA repair; apoptosis
Li-Fraumeni syndrome; mutated in >50% of all sporadic cancers
RB
G1/S checkpoint control (binds E2F transcription factor)
Retinoblastoma; osteosarcoma
APC
Negative regulator of WNT/β-catenin signaling
Familial adenomatous polyposis (FAP); sporadic colon cancer
BRCA1/BRCA2
DNA double-strand break repair (homologous recombination)
Hereditary breast/ovarian cancer; BRCA2 also pancreatic, prostate
VHL
Degradation of HIF-1α (prevents angiogenesis signaling under normoxia)
von Hippel-Lindau syndrome (renal cell carcinoma, hemangioblastoma, pheochromocytoma)
WT1
Transcription factor (kidney development)
Wilms tumor (nephroblastoma)
NF1
GAP protein (inactivates RAS)
Neurofibromatosis type 1 (neurofibromas, optic glioma, café-au-lait spots)
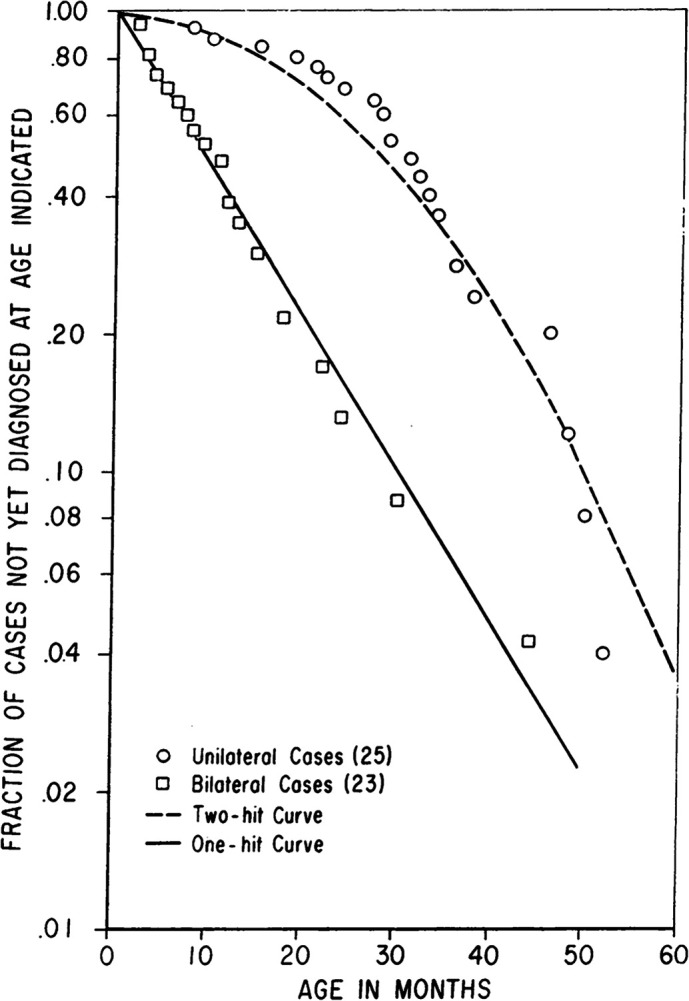
Figure 30 — Knudson’s Two-Hit Hypothesis. The original statistical analysis that led Knudson to propose the two-hit hypothesis. In hereditary retinoblastoma (one germline hit), a single somatic mutation suffices, producing the straight line. In sporadic cases (two somatic hits required), the age-incidence curve is hyperbolic, reflecting the lower probability of two independent mutations occurring in the same cell.
Knudson’s two-hit hypothesis: both alleles of a tumor suppressor must be inactivated for loss of function. In hereditary cancers (e.g., retinoblastoma), one hit is inherited (germline mutation) and only one somatic hit is needed — explaining earlier onset and bilateral/multifocal tumors. In sporadic cases, both hits must occur somatically in the same cell, which is much less likely and occurs later in life.
Chemical & Radiation Carcinogenesis
Carcinogen
Target / Mechanism
Associated Cancer
Aflatoxin B1 (Aspergillus flavus/parasiticus)
p53 mutation (codon 249, G→T transversion)
Hepatocellular carcinoma (synergistic with HBV)
Asbestos
Chronic inflammation; direct mesothelial cell toxicity
Mesothelioma (pleural); bronchogenic carcinoma (synergistic with smoking)
Antibodies against presynaptic voltage-gated Ca2+ channels at NMJ
SCLC
Trousseau syndrome
Migratory superficial thrombophlebitis; hypercoagulable state
Pancreatic adenocarcinoma; other mucin-secreting cancers
Acanthosis nigricans
Insulin-like growth factors from tumor
Gastric adenocarcinoma
Dermatomyositis
Autoimmune; immune cross-reactivity
Ovarian, lung, gastric cancers
Limbic encephalitis
Anti-Hu antibodies (anti-neuronal)
SCLC
Cerebellar degeneration
Anti-Yo antibodies (anti-Purkinje cell)
Ovarian, breast
Small cell lung carcinoma (SCLC) is the most common cancer associated with paraneoplastic syndromes, including SIADH, ectopic ACTH/Cushing, and Lambert-Eaton syndrome. SCLC is a neuroendocrine tumor with the ability to produce diverse peptide hormones. Always consider occult malignancy in a patient presenting with unexplained endocrine or neurologic syndromes.
22 Hypersensitivity Reactions (Types I–IV)
Hypersensitivity reactions are exaggerated or inappropriate immune responses to antigens that result in tissue damage. They are classified by the Gell and Coombs system into four types.
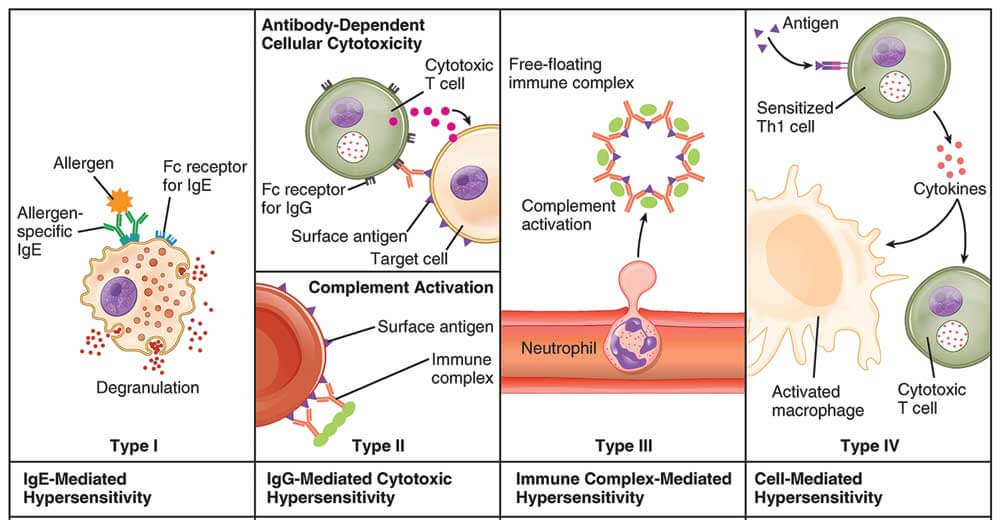
Figure 31 — Types of Hypersensitivity Reactions. The Gell and Coombs classification of hypersensitivity: Type I (IgE-mediated immediate), Type II (antibody-mediated cytotoxic), Type III (immune complex-mediated), and Type IV (T cell-mediated delayed). Each type has a distinct immunologic mechanism and clinical time course.
Classification of Hypersensitivity
Type
Name
Mechanism
Timing
Classic Examples
I
Immediate (anaphylactic)
Preformed IgE on mast cells/basophils; cross-linking by antigen → degranulation (histamine, leukotrienes, prostaglandins)
Late phase (6–24 hours): Recruitment of eosinophils, basophils, TH2 cells → sustained inflammation; responsible for the “second wave” of symptoms in asthma
Serum tryptase is the best confirmatory test for anaphylaxis; it peaks 1–2 hours after onset and remains elevated for several hours. Total IgE and specific IgE (RAST/ImmunoCAP) confirm atopic sensitization but do not confirm clinical allergy. Skin prick testing remains the most sensitive in vivo test for IgE-mediated allergy.
23 Autoimmune Disease & Transplant Rejection
Mechanisms of Autoimmunity
Autoimmune disease results from failure of self-tolerance — the immune system attacks the body’s own tissues. Central tolerance (deletion of self-reactive T and B cells in thymus and bone marrow) and peripheral tolerance (anergy, regulatory T cells, apoptosis of self-reactive lymphocytes) normally prevent autoimmunity. Breakdown of these mechanisms, often in genetically susceptible individuals (HLA associations), leads to autoimmune disease.
HLA Associations in Autoimmune Disease
HLA Allele
Disease
Relative Risk
HLA-B27
Ankylosing spondylitis
~90×
HLA-B27
Reactive arthritis (Reiter syndrome)
~40×
HLA-DR4
Rheumatoid arthritis
~6×
HLA-DR3/DR4
Type 1 diabetes mellitus
~20×
HLA-DR2
Multiple sclerosis; Goodpasture syndrome; SLE
Variable
HLA-DQ2/DQ8
Celiac disease
>95% carry DQ2 or DQ8
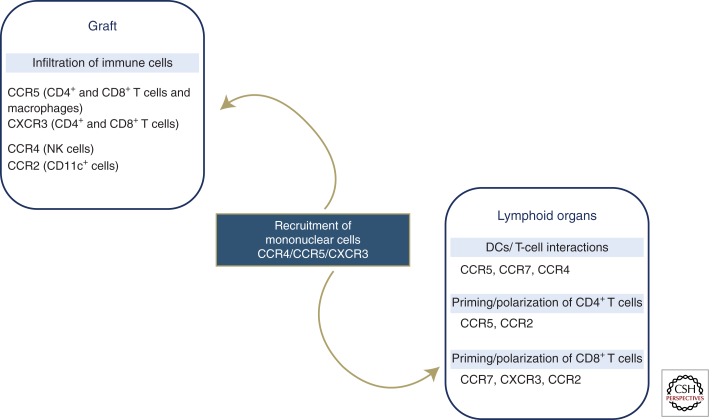
Figure 32 — Transplant Rejection Mechanisms. Immunologic mechanisms involved in graft rejection, showing chemokine-mediated recruitment of immune effector cells to the transplanted organ. Host T cells recognize donor MHC antigens through direct and indirect pathways, leading to cellular and humoral rejection.
Graft-versus-host disease (GVHD) occurs when immunocompetent donor T cells in a bone marrow transplant attack immunocompromised host tissues. Target organs: skin (dermatitis), liver (jaundice), GI tract (diarrhea). Acute GVHD occurs within 100 days; chronic GVHD after 100 days with features resembling autoimmune disease (scleroderma-like skin, sicca syndrome).
Key Autoantibody Associations
Autoantibody
Disease
ANA (antinuclear antibody)
Sensitive (but not specific) for SLE; also positive in drug-induced lupus, scleroderma, Sjögren
Anti-dsDNA
Specific for SLE; correlates with disease activity and lupus nephritis
Antiphospholipid syndrome: recurrent thrombosis, pregnancy loss; paradoxically prolongs aPTT in vitro but causes thrombosis in vivo
24 Amyloidosis
Amyloidosis is a group of disorders characterized by extracellular deposition of misfolded proteins in an abnormal fibrillar configuration (cross-beta-pleated sheet). These deposits are insoluble, resistant to proteolysis, and progressively damage organs by displacing normal parenchyma.
Diagnosis
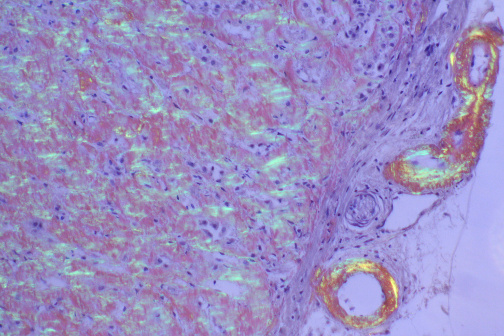
Amyloid deposits stain with Congo red and display apple-green birefringence under polarized light. This is the gold standard histologic test. Tissue can be obtained from abdominal fat pad aspirate, rectal biopsy, or affected organ biopsy.
Figure 33 — Amyloid Deposits (Congo Red Stain). Histologic demonstration of amyloid deposits using Congo red stain. Under polarized light, amyloid displays characteristic apple-green birefringence due to the ordered cross-beta-pleated sheet configuration of the misfolded fibrillar proteins. This is the gold standard histologic test for amyloidosis.
Types of Amyloidosis
Type
Precursor Protein
Fibril Protein
Associated Condition
Organs Affected
AL (primary)
Immunoglobulin light chains
AL
Plasma cell dyscrasias (multiple myeloma, Waldenström); B cell lymphomas
The heart is the most important organ involved in AL amyloidosis and is the leading cause of death. Presents as restrictive cardiomyopathy with diastolic dysfunction, thick ventricular walls (but NOT hypertrophy — infiltration), low voltage on ECG (paradox with thick walls on echo), and heart failure. Senile cardiac amyloidosis (wild-type ATTR) is increasingly recognized in elderly patients with HFpEF and can now be diagnosed non-invasively with technetium pyrophosphate (Tc-PYP) scan. Treatment with tafamidis (TTR stabilizer) reduces mortality in ATTR cardiac amyloidosis.
25 Genetic Disorders & Inheritance Patterns
Autosomal Dominant Disorders
One mutant allele is sufficient to cause disease. Affected individuals typically have one affected parent. Variable expressivity and incomplete penetrance are common. Key examples:
Fragile X syndrome — CGG repeat expansion in FMR1 → intellectual disability, long face, large ears, macroorchidism; most common inherited cause of intellectual disability
Chromosomal Disorders
Disorder
Karyotype
Key Features
Down syndrome (Trisomy 21)
47,XX/XY,+21
Most common viable autosomal trisomy; intellectual disability; flat facies; epicanthal folds; simian crease; duodenal atresia; Hirschsprung disease; AV canal defect; increased risk of ALL and early-onset Alzheimer (APP gene on chr 21); maternal age >35 is major risk factor
Edwards syndrome (Trisomy 18)
47,XX/XY,+18
Severe intellectual disability; rocker-bottom feet; clenched fists (overlapping fingers); micrognathia; congenital heart defects; most die within 1 year
Patau syndrome (Trisomy 13)
47,XX/XY,+13
Holoprosencephaly; cleft lip/palate; polydactyly; microphthalmia; congenital heart defects; cutis aplasia; most die within 1 year
Turner syndrome
45,X
Short stature; shield chest; webbed neck; lymphedema of hands/feet at birth; streak gonads; coarctation of aorta; horseshoe kidney; no intellectual disability; most common cause of primary amenorrhea
Klinefelter syndrome
47,XXY
Tall stature; gynecomastia; small firm testes; infertility (azoospermia); ↑ FSH/LH; female pattern hair distribution; increased risk of breast cancer and SLE
Trinucleotide Repeat Disorders
Disease
Repeat
Gene
Key Feature
Huntington
CAG
HTT (chr 4)
Anticipation (paternal transmission)
Fragile X
CGG
FMR1 (X-linked)
Anticipation (maternal transmission)
Myotonic dystrophy
CTG
DMPK (chr 19)
Most common adult muscular dystrophy; myotonia; cataracts
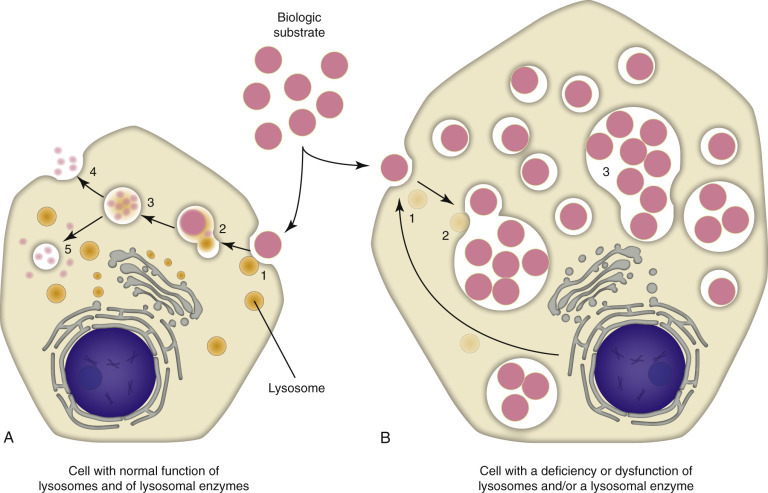
Lysosomal storage diseases result from inherited deficiency of specific lysosomal enzymes, leading to accumulation of undigested substrates within lysosomes. Most are autosomal recessive.
Lysosomal Storage Diseases
Disease
Enzyme Deficiency
Accumulated Substrate
Key Features
Tay-Sachs
Hexosaminidase A
GM2 ganglioside
Cherry-red spot on macula; progressive neurodegeneration; death by age 3; common in Ashkenazi Jewish; NO hepatosplenomegaly (distinguishes from Niemann-Pick)
X-linked recessive; similar to Hurler but milder; NO corneal clouding; aggressive behavior
Figure 34 — Lysosomal Storage Diseases. Schematic overview of lysosomal storage diseases showing the enzymatic defects and accumulated substrates. Each disease results from deficiency of a specific lysosomal enzyme, leading to progressive accumulation of undigested substrates within lysosomes and characteristic clinical manifestations.
Glycogen Storage Diseases
Type
Disease
Enzyme Deficiency
Key Features
I
Von Gierke
Glucose-6-phosphatase
Severe fasting hypoglycemia; hepatomegaly; lactic acidosis; hyperuricemia; hyperlipidemia
II
Pompe
Acid maltase (α-1,4-glucosidase) — lysosomal
Cardiomegaly (most prominent); hypotonia; early death (infantile form); only GSD that is a lysosomal storage disease
III
Cori (Forbes)
Debranching enzyme
Similar to Von Gierke but milder; gluconeogenesis intact
V
McArdle
Muscle glycogen phosphorylase
Exercise intolerance; myoglobinuria; no rise in blood lactate with exercise; “second wind” phenomenon
Mnemonics for lysosomal storage diseases: “Tay-Sachs lacks heXosaminidase” (X for hex); “Niemann-Pick has No sphingomyelinase”; “Gaucher has Glucocerebrosidase deficiency” (G for G). Remember that Fabry and Hunter are the only X-linked lysosomal storage diseases (“Fabulous Hunters are X-linked”).
27 Organ System Pathology Overview
General pathology principles recur across organ systems. This section provides a rapid-reference map linking pathophysiologic mechanisms to their most important organ-specific manifestations.
MEN 1 (3 P’s: pituitary, parathyroid, pancreas); MEN 2A (medullary thyroid CA, pheo, parathyroid hyperplasia; RET mutation); MEN 2B (medullary thyroid CA, pheo, mucosal neuromas, marfanoid habitus)
Multiple sclerosis (Type IV hypersensitivity; oligoclonal bands in CSF; periventricular plaques); Guillain-Barré (ascending paralysis; anti-ganglioside antibodies; albuminocytologic dissociation)
Neurodegenerative
Alzheimer (Aβ plaques + neurofibrillary tangles of hyperphosphorylated tau); Parkinson (loss of dopaminergic neurons in substantia nigra; Lewy bodies = α-synuclein)
Neoplastic
Glioblastoma (most common primary brain tumor in adults; GBM = grade IV; pseudopalisading necrosis); meningioma (2nd most common; dural-based; psammoma bodies); schwannoma (S-100+; CN VIII → acoustic neuroma)
A systematic approach to organ pathology applies the general pathology framework: for any organ, consider (1) vascular/hemodynamic causes, (2) inflammatory/infectious causes, (3) neoplastic causes, (4) degenerative/metabolic causes, and (5) genetic/developmental causes. This exhaustive list prevents you from missing diagnoses on differential.
28 High-Yield Review & Board Pearls
CELL INJURY & DEATH
Most common cause of cell injury: hypoxia/ischemia
First biochemical change in ischemia: decreased oxidative phosphorylation → ATP depletion
First morphologic change in reversible injury: cellular swelling (hydropic change)
Most reliable markers of irreversible injury: mitochondrial dense amorphous densities + plasma membrane disruption
Hallmark of necrosis: inflammation; hallmark of apoptosis: no inflammation
Brain infarcts: liquefactive necrosis (exception to solid organ rule)
Caseous necrosis: think tuberculosis
Fat necrosis with saponification: think acute pancreatitis
Fibrinoid necrosis in vessel walls: think malignant hypertension or vasculitis
INFLAMMATION
First cells to arrive in acute inflammation: neutrophils (peak 6–24 hours)
Most important cell in chronic inflammation: macrophage
Most important cell in wound healing: macrophage
Most important mediator of fever: PGE2 (produced in hypothalamus in response to IL-1, TNF, IL-6)
C5a: most potent chemotactic factor for neutrophils (also anaphylatoxin)
LTB4: potent neutrophil chemotaxis
LTC4/D4/E4: bronchoconstriction (slow-reacting substances of anaphylaxis)
Sickle cell trait (HbAS): protective against Plasmodium falciparum malaria
Autoantibody specificity: anti-dsDNA = lupus nephritis activity; anti-Smith = most specific for SLE; anti-CCP = most specific for RA
Antiphospholipid syndrome: prolonged aPTT but thrombosis in vivo (not bleeding)
Board Strategy: Pathophysiology is the highest-yield subject for USMLE Step 1. For each disease, know the mechanism (etiology + pathogenesis), the expected morphologic changes (gross and microscopic), and the clinical consequences (symptoms, lab findings, complications). Questions typically present a clinical vignette and ask you to identify the underlying mechanism or predict the next step in the disease process. Recognize patterns: all forms of necrosis, the cardinal features of each type of hypersensitivity, Virchow’s triad, and the hallmarks of cancer are tested repeatedly.