Physiology
Cardiovascular, respiratory, renal, GI, neurophysiology, endocrine, acid-base, and every physiologic principle, equation, regulatory mechanism, and clinical correlation across the full scope of human physiology.
01 Overview & Scope of Physiology
Physiology is the study of how living organisms function — from the molecular events within a single ion channel to the integrated responses of the whole body during exercise, hemorrhage, or disease. It bridges the gap between the structural knowledge of anatomy and histology and the clinical reasoning required at the bedside. Understanding normal physiology is the prerequisite for understanding pathophysiology, pharmacology, and every clinical specialty.
Every disease is, at its core, a disruption of normal physiology. A clinician who understands Starling forces can predict the fluid shifts in heart failure, nephrotic syndrome, and cirrhosis. One who understands the oxygen-hemoglobin dissociation curve can anticipate how acidosis, anemia, or carbon monoxide poisoning will affect tissue oxygenation. Physiology is the language of clinical medicine.
Levels of Organization
Physiology operates at every scale: molecular (ion channels, enzyme kinetics, receptor-ligand interactions), cellular (membrane transport, signal transduction, gene expression), tissue/organ (cardiac contractility, renal filtration, neural circuits), and systems (cardiovascular regulation of blood pressure, respiratory gas exchange, endocrine feedback loops). Integration across these levels produces homeostasis — the maintenance of a stable internal environment despite constant external perturbation.
Core Disciplines Within Physiology
| Discipline | Focus | Key Concepts |
|---|---|---|
| Cardiovascular | Heart pump function, vascular hemodynamics | Cardiac output, Starling law, MAP, baroreflexes |
| Respiratory | Gas exchange, ventilation mechanics | V/Q matching, compliance, O2-Hb curve, alveolar gas equation |
| Renal | Filtration, electrolyte/water balance | GFR, clearance, RAAS, countercurrent multiplier |
| GI | Motility, secretion, absorption | Migrating motor complex, bile acid circulation, nutrient transporters |
| Neurophysiology | Electrical signaling, synaptic transmission | Action potential, neurotransmitters, sensory/motor integration |
| Endocrine | Hormone synthesis, feedback loops | HPA axis, thyroid axis, insulin/glucagon, calcium homeostasis |
| Musculoskeletal | Contraction, motor unit recruitment | Sliding filament theory, excitation-contraction coupling |
02 Cell Physiology & Membrane Transport
All physiologic processes begin at the cell membrane — a 7-8 nm phospholipid bilayer that separates the intracellular from the extracellular environment and controls the selective passage of ions, nutrients, and signaling molecules.
Membrane Transport Mechanisms
| Mechanism | Energy Source | Direction | Examples |
|---|---|---|---|
| Simple diffusion | None (passive) | Down gradient | O2, CO2, steroid hormones, fatty acids |
| Facilitated diffusion | None (passive) | Down gradient via carrier/channel | GLUT transporters (glucose into cells), ion channels |
| Primary active transport | ATP hydrolysis | Against gradient | Na+/K+-ATPase, Ca2+-ATPase, H+/K+-ATPase |
| Secondary active transport | Ion gradient (created by primary) | Against gradient (one solute) | SGLT (Na+/glucose cotransport), Na+/H+ exchanger |
| Osmosis | None (passive) | Water follows solute gradient | Aquaporins (AQP-2 in collecting duct, regulated by ADH) |
| Endocytosis/Exocytosis | ATP | Bulk transport | Receptor-mediated endocytosis (LDL), neurotransmitter release |
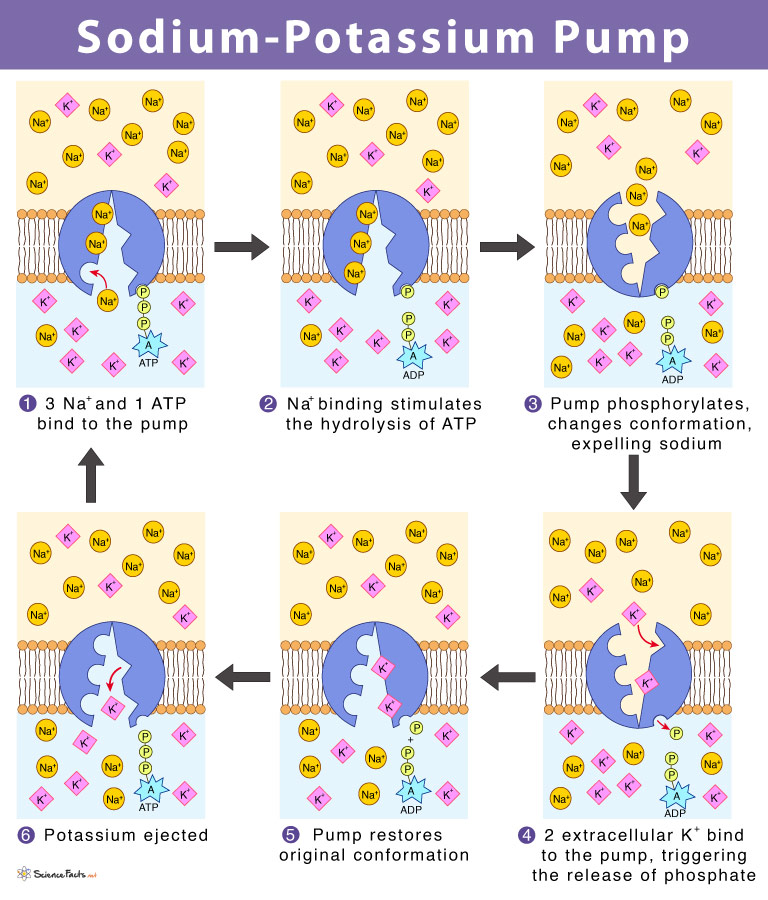
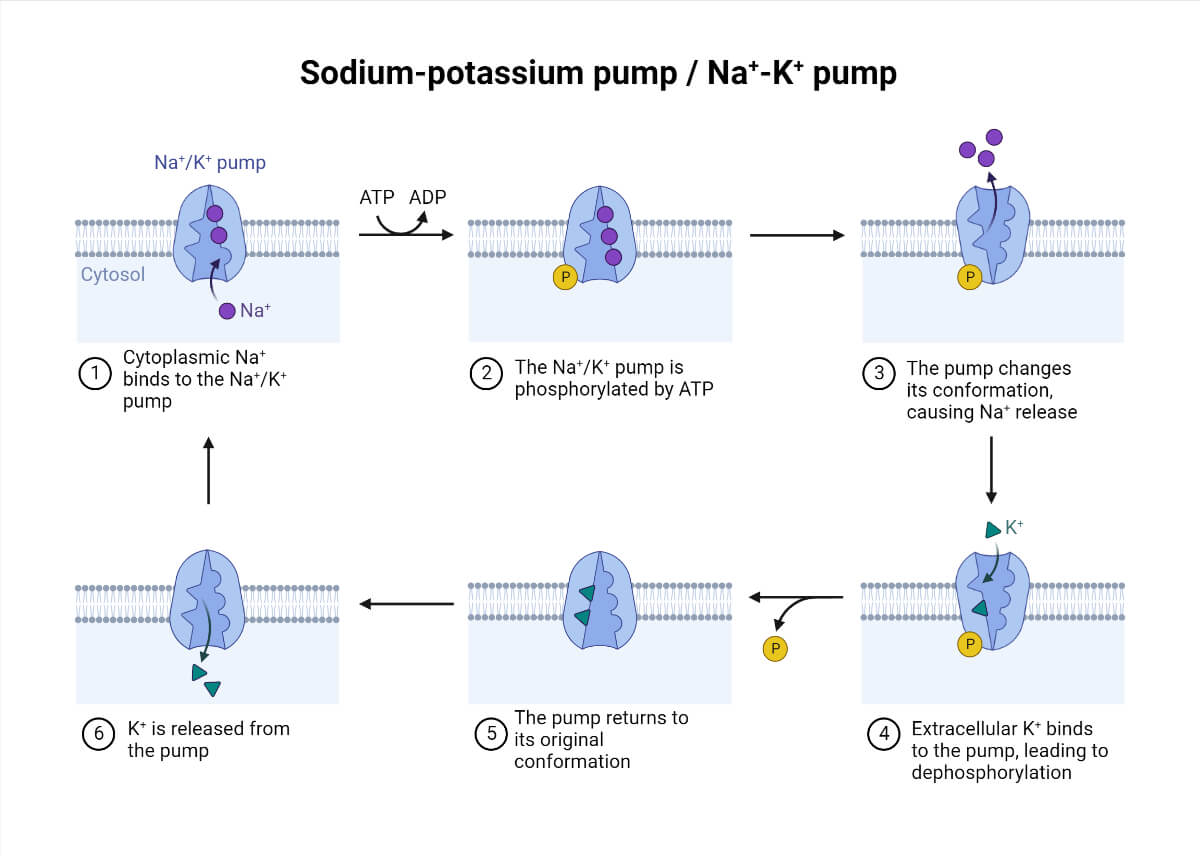
The Na+/K+-ATPase
The single most important membrane pump in human physiology. It transports 3 Na+ out and 2 K+ in per ATP hydrolyzed, maintaining the electrochemical gradients that drive secondary active transport, set the resting membrane potential, and regulate cell volume. It consumes approximately 30% of total body ATP at rest. It is inhibited by cardiac glycosides (digoxin, ouabain) — this increases intracellular Na+, which in turn reduces Ca2+ extrusion via the Na+/Ca2+ exchanger, increasing intracellular Ca2+ and enhancing cardiac contractility (the basis of digoxin's inotropic effect).
Signal Transduction Fundamentals
Cell signaling follows a general paradigm: ligand binds receptor → intracellular signal cascade → cellular response. Receptors fall into four major categories: (1) Ion channel-linked (ligand-gated ion channels — nicotinic ACh receptor, GABA-A, NMDA): fastest response, milliseconds. (2) G-protein-coupled receptors (GPCRs): the largest family (~800 in the human genome), including adrenergic, muscarinic, opioid, and most hormone receptors; response in seconds to minutes via cAMP, IP3/DAG, or ion channel modulation. (3) Enzyme-linked receptors (receptor tyrosine kinases — insulin receptor, growth factor receptors): response in minutes to hours via phosphorylation cascades (RAS-MAP kinase, PI3K-Akt). (4) Intracellular/nuclear receptors (steroid hormones, thyroid hormones, vitamin D, retinoids): slowest response, hours to days, via direct gene transcription regulation.
G-Protein Signaling Pathways
| G-Protein | Effector | Second Messenger | Receptors Using This Pathway |
|---|---|---|---|
| Gs | Stimulates adenylyl cyclase | Increased cAMP → PKA | Beta-1, Beta-2, D1, H2, V2, ACTH-R, TSH-R, LH-R, FSH-R, PTH-R, glucagon-R |
| Gi | Inhibits adenylyl cyclase | Decreased cAMP | Alpha-2, M2, D2, opioid receptors, somatostatin-R |
| Gq | Activates PLC | IP3 (→ Ca2+ release) + DAG (→ PKC) | Alpha-1, M1, M3, H1, V1, AT1, 5-HT2, GnRH-R, TRH-R, oxytocin-R |
| Gt (transducin) | Activates PDE | Decreased cGMP | Rhodopsin (phototransduction) |
Resting Membrane Potential
The resting membrane potential of most cells is approximately -70 to -90 mV (inside negative relative to outside). It is determined primarily by the K+ equilibrium potential (EK approximately -90 mV) because the resting membrane is most permeable to K+ through leak channels. The Nernst equation calculates the equilibrium potential for a single ion:
Eion = (61.5 / z) x log([ion]outside / [ion]inside)
At 37 degrees C; z = valence of the ion
EK = 61.5 x log(4/140) approximately -94 mV
ENa = 61.5 x log(140/14) approximately +61 mV
The Goldman-Hodgkin-Katz (GHK) equation accounts for multiple ion permeabilities simultaneously and more accurately predicts the actual resting potential, which lies between EK and ENa, closer to EK because K+ permeability dominates at rest. During an action potential, Na+ permeability transiently dominates, driving the membrane toward ENa (+61 mV).
Ion Composition: Intracellular vs Extracellular
| Ion | Intracellular (mEq/L) | Extracellular (mEq/L) | Eion (mV) |
|---|---|---|---|
| Na+ | 14 | 140 | +61 |
| K+ | 140 | 4 | -94 |
| Ca2+ | 0.0001 | 2.5 | +132 |
| Cl- | 4 | 105 | -89 |
03 Homeostasis & Feedback Systems
Homeostasis — the maintenance of a stable internal environment — is the central organizing principle of physiology. The body continuously monitors variables (temperature, pH, osmolality, blood glucose, blood pressure) and activates corrective responses through feedback loops.
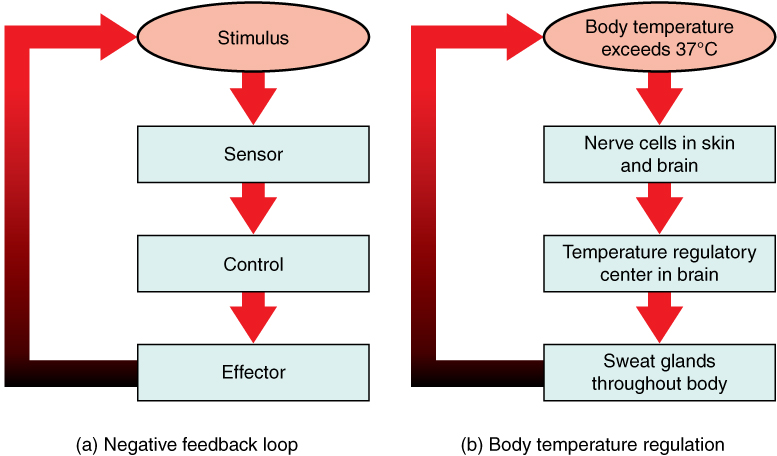
Negative Feedback
The dominant regulatory mechanism in physiology. A change in a variable triggers a response that opposes the change and returns the variable toward its set point. Examples include: thermoregulation (increased body temperature triggers sweating and vasodilation to reduce temperature); blood glucose regulation (hyperglycemia stimulates insulin secretion, which lowers glucose); blood pressure regulation (baroreceptors detect elevated BP and increase vagal tone to lower heart rate and BP); thyroid axis (elevated T3/T4 suppresses TSH release from the anterior pituitary).
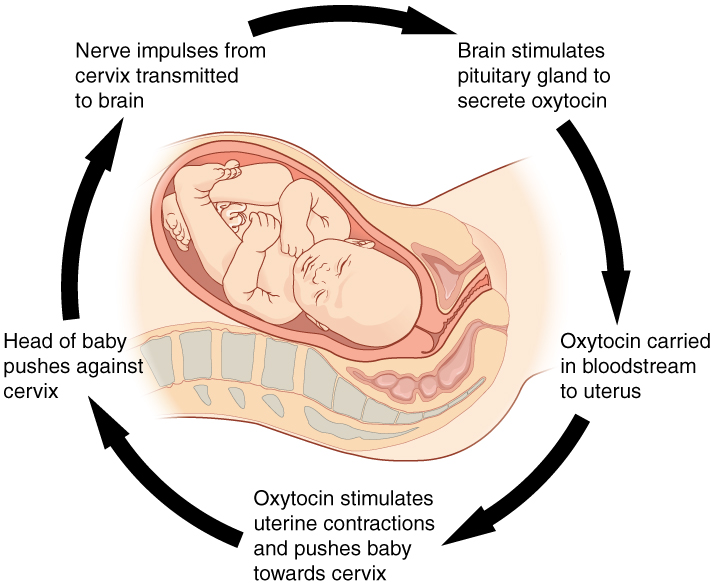
Positive Feedback
A change in a variable triggers a response that amplifies the change. These are inherently unstable and drive the system to completion. Physiologic examples: oxytocin during labor (uterine contractions stimulate oxytocin release, which causes stronger contractions until delivery); the LH surge in ovulation (rising estrogen triggers a positive feedback surge of LH that causes follicular rupture); the upstroke of the action potential (Na+ influx depolarizes the membrane, opening more Na+ channels); blood clotting cascade (thrombin activates more prothrombin in an amplifying cascade).
Pathologic positive feedback produces vicious cycles: hemorrhagic shock (decreased cardiac output causes tissue ischemia, which impairs cardiac function, further decreasing output); DIC (clot consumption leads to bleeding, which triggers more clotting and consumption). Recognizing and breaking these cycles is the basis of emergency resuscitation.
Feed-Forward Control
Anticipatory regulation that activates responses before the change occurs. The cephalic phase of gastric secretion (seeing or smelling food triggers vagal stimulation of acid and enzyme release before food reaches the stomach) and pre-exercise tachycardia (sympathetic activation increases heart rate before exercise begins) are classic examples.
Normal Physiologic Set Points
| Variable | Normal Range | Primary Regulatory Mechanism |
|---|---|---|
| Core body temperature | 36.5-37.5 degrees C | Hypothalamic thermostat, sweating/shivering |
| Arterial pH | 7.35-7.45 | Bicarbonate buffer, respiratory compensation, renal compensation |
| Plasma osmolality | 280-295 mOsm/kg | ADH release/suppression, thirst |
| Serum Na+ | 135-145 mEq/L | ADH, aldosterone, thirst |
| Serum K+ | 3.5-5.0 mEq/L | Aldosterone, insulin, pH shifts |
| Fasting blood glucose | 70-100 mg/dL | Insulin/glucagon balance |
| Mean arterial pressure | 70-105 mmHg | Baroreceptors, RAAS, sympathetic tone |
| PaCO2 | 35-45 mmHg | Central/peripheral chemoreceptors adjusting ventilation |
| Serum Ca2+ (ionized) | 4.5-5.5 mg/dL | PTH, vitamin D, calcitonin |
04 Cardiac Electrophysiology
The heart is a self-excitable organ. Specialized pacemaker cells generate spontaneous depolarizations that propagate through a precisely organized conduction system, ensuring coordinated atrial and ventricular contraction roughly 100,000 times per day. Understanding cardiac electrophysiology is essential for interpreting ECGs, managing arrhythmias, and understanding the mechanisms of antiarrhythmic drugs.
The cardiac action potential differs fundamentally from the nerve/skeletal muscle action potential. The plateau phase (phase 2), mediated by L-type Ca2+ channels, prolongs the cardiac AP to ~250-300 ms (vs ~1-2 ms in nerve). This ensures: (1) a long refractory period preventing tetanic contraction, (2) adequate Ca2+ entry to trigger Ca2+-induced Ca2+ release from the SR, and (3) time for complete ejection before relaxation begins.
Electrical Properties of the Heart
The heart possesses five key electrical properties: Automaticity (ability to spontaneously depolarize — SA node, AV node, Purkinje fibers), Rhythmicity (regularity of depolarization), Conductivity (ability to propagate impulses cell-to-cell via gap junctions in intercalated discs), Excitability (ability to respond to a stimulus), and Contractility (ability to generate force). The hierarchy of pacemakers ensures a backup if the SA node fails: SA node (60-100 bpm) → AV node (40-60 bpm) → ventricular/Purkinje (20-40 bpm). If the SA node fails, the AV node takes over (junctional rhythm); if both fail, the ventricles generate an escape rhythm at 20-40 bpm — often insufficient to maintain adequate cardiac output.
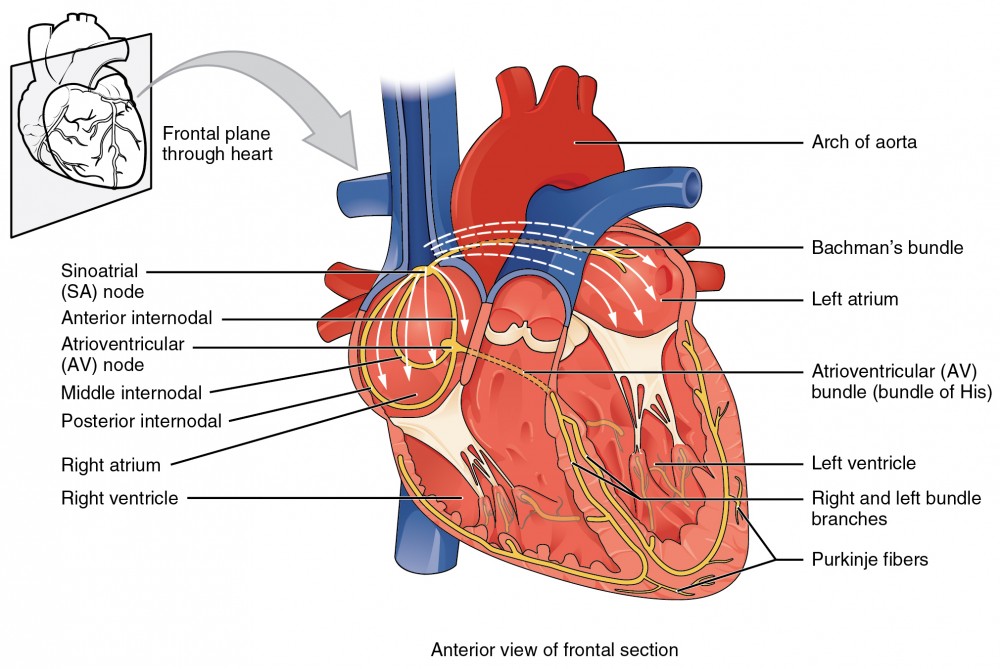
The Conduction System
The SA node (right atrium, near the SVC junction) is the primary pacemaker with an intrinsic rate of 60-100 bpm. Impulses travel through atrial myocardium (internodal pathways) to the AV node (right atrial septum, near the coronary sinus) — the only normal electrical connection between atria and ventricles, with intrinsic rate 40-60 bpm. The AV node introduces a critical 0.1-second delay that allows atrial contraction to complete before ventricular systole begins (the "atrial kick"). From the AV node, the signal passes through the Bundle of His → right and left bundle branches → Purkinje fibers (intrinsic rate 20-40 bpm), which rapidly depolarize the ventricular myocardium from endocardium to epicardium.
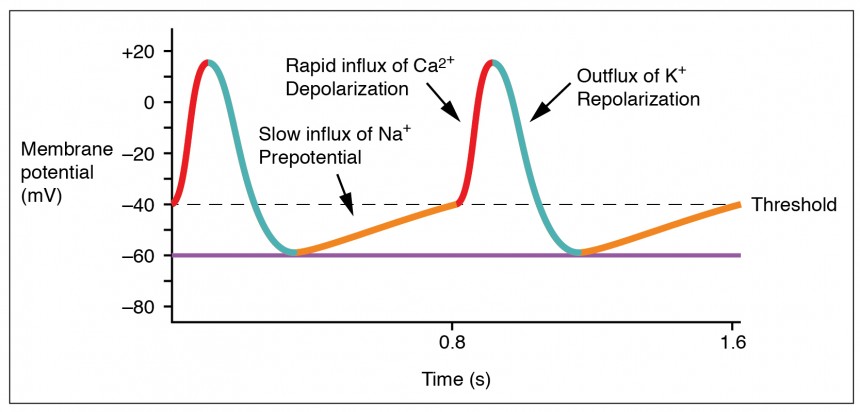
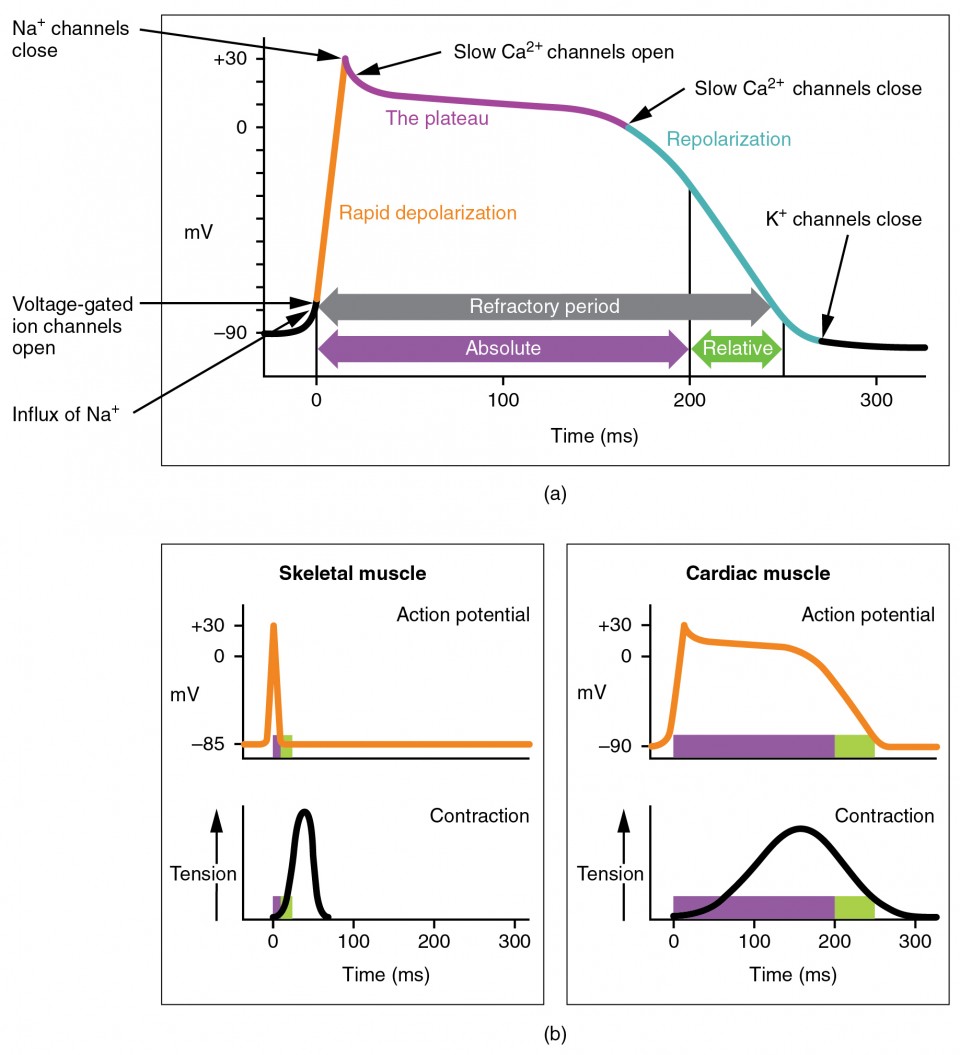
Action Potentials
| Phase | Ventricular Myocyte | Pacemaker Cell (SA/AV) |
|---|---|---|
| Phase 0 (rapid depolarization) | Fast Na+ influx (INa) | Slow Ca2+ influx (ICa-L) — no fast Na+ channels |
| Phase 1 (early repolarization) | Transient K+ efflux (Ito) | Absent |
| Phase 2 (plateau) | Ca2+ influx (ICa-L) balanced by K+ efflux | Absent |
| Phase 3 (repolarization) | K+ efflux (IKr, IKs) | K+ efflux (IK) |
| Phase 4 (resting/diastolic) | Stable at -90 mV (IK1) | Slow spontaneous depolarization via "funny current" (If, Na+ influx) + ICa-T |
Clinical Correlations — Electrophysiology
Class I antiarrhythmics (Na+ channel blockers): slow phase 0 in ventricular myocytes. Class III antiarrhythmics (K+ channel blockers, e.g., amiodarone, sotalol): prolong phase 3, prolonging QT interval. Class IV (Ca2+ channel blockers, e.g., verapamil, diltiazem): slow phase 0 in pacemaker cells, useful for SVT and rate control of atrial fibrillation. Ivabradine: selectively blocks If current, reducing SA node firing rate without affecting contractility — used in heart failure with elevated resting heart rate.
Hyperkalemia reduces the K+ gradient, partially depolarizing the resting membrane potential. This initially increases excitability (peaked T waves) but progressively inactivates Na+ channels, slowing conduction (widened QRS) and eventually causing sine wave pattern and cardiac arrest. Calcium gluconate stabilizes the membrane; insulin/glucose and beta-agonists shift K+ intracellularly.
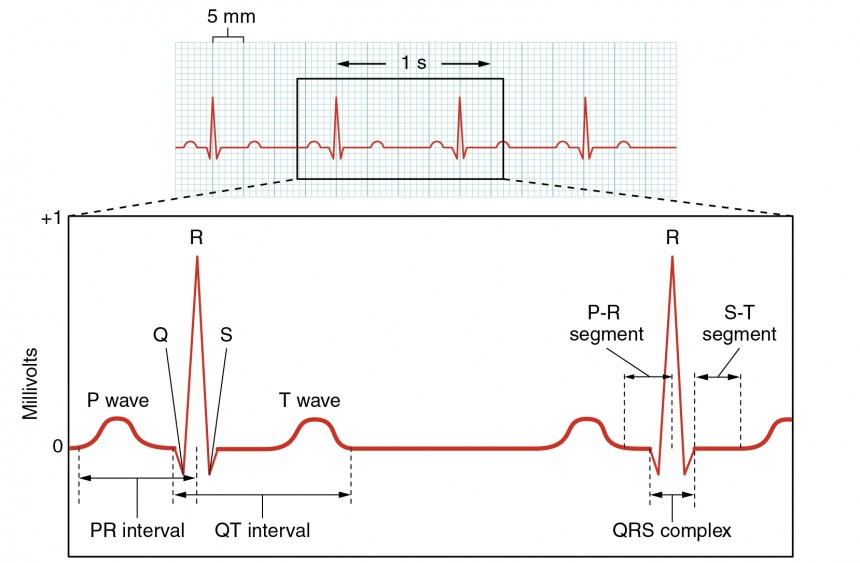
ECG Correlations to Cardiac Electrophysiology
| ECG Component | Electrophysiologic Event | Duration | Clinical Significance |
|---|---|---|---|
| P wave | Atrial depolarization | < 120 ms | Absent in atrial fibrillation; peaked in right atrial enlargement; notched in left atrial enlargement |
| PR interval | Atrial depolarization + AV nodal delay | 120-200 ms | Prolonged in 1st degree AV block; shortened in WPW (delta wave) |
| QRS complex | Ventricular depolarization | < 120 ms | Widened in bundle branch block, hyperkalemia, or ventricular rhythm |
| ST segment | Ventricular plateau (phase 2) | Variable | Elevation = STEMI, pericarditis; depression = ischemia, digoxin effect |
| T wave | Ventricular repolarization | Variable | Peaked in hyperkalemia; inverted in ischemia, strain, PE |
| QT interval | Total ventricular depolarization + repolarization | QTc < 470 ms (F), < 450 ms (M) | Prolonged QT → risk of Torsades de Pointes (polymorphic VT) |
05 The Cardiac Cycle & Pressure-Volume Loops
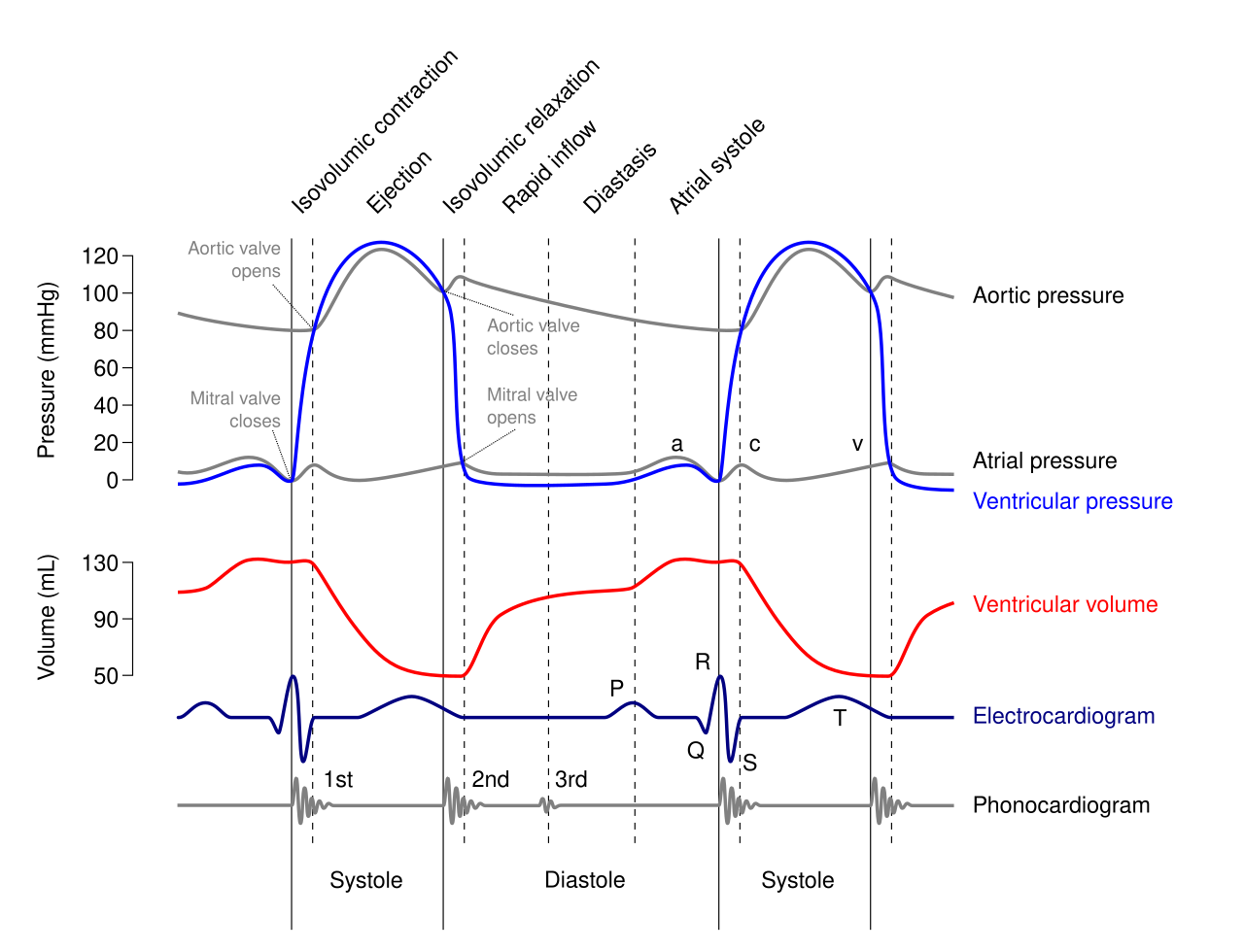
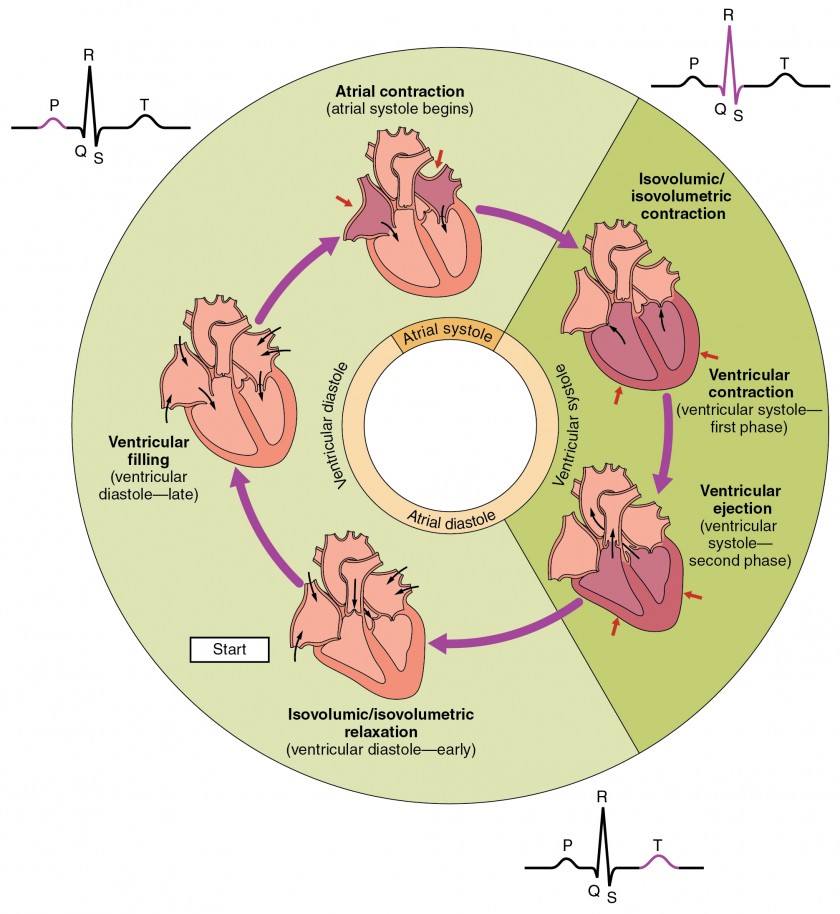
Phases of the Cardiac Cycle
| Phase | Valves | Events |
|---|---|---|
| Isovolumetric contraction | All closed | LV pressure rises; volume constant; begins at mitral valve closure (S1) |
| Rapid ejection | Aortic opens | LV pressure exceeds aortic pressure; peak aortic flow |
| Reduced ejection | Aortic open | LV and aortic pressures decline; aortic pressure may briefly exceed LV |
| Isovolumetric relaxation | All closed | LV pressure falls; volume constant; begins at aortic valve closure (S2) |
| Rapid filling | Mitral opens | Passive filling (~80% of filling); S3 if pathologic |
| Atrial contraction | Mitral open | Active "atrial kick" (~20% of filling); S4 if stiff ventricle |
Heart Sounds
S1: closure of mitral and tricuspid valves (beginning of systole). S2: closure of aortic and pulmonic valves (beginning of diastole). Physiologic splitting of S2: inspiration increases venous return to the right heart, delaying pulmonic valve closure — A2 then P2 heard during inspiration. Fixed splitting: ASD (constant left-to-right shunt equalizes inspiratory/expiratory volumes). Paradoxical splitting: LBBB or aortic stenosis delays aortic valve closure — P2 then A2, splitting on expiration.
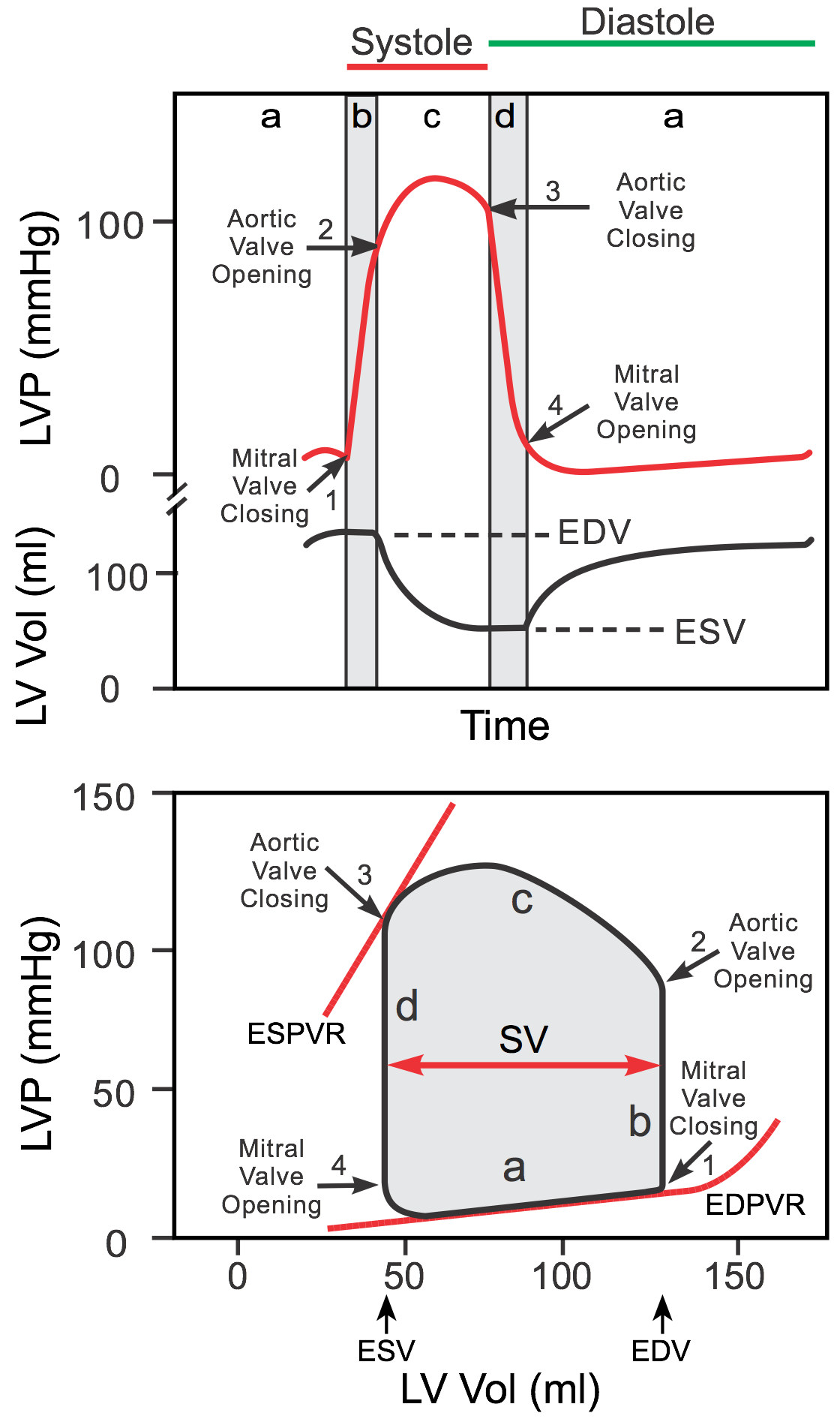
Pressure-Volume (PV) Loops
The PV loop is the most powerful tool for understanding cardiac mechanics. The loop plots LV pressure (y-axis) against LV volume (x-axis) over one cardiac cycle, proceeding counterclockwise. The area within the loop represents stroke work.
Bottom-right: End-diastolic volume (EDV, preload) — point of mitral valve closure.
Top-right: End of isovolumetric contraction — aortic valve opens.
Top-left: End-systolic volume (ESV) — aortic valve closes.
Bottom-left: End of isovolumetric relaxation — mitral valve opens.
ESPVR (end-systolic PV relationship): upper-left boundary, reflects contractility (slope = Ees).
EDPVR (end-diastolic PV relationship): lower boundary, reflects compliance.
PV Loop Changes in Disease
| Condition | PV Loop Change | Mechanism |
|---|---|---|
| Increased preload | Loop shifts right; wider loop; increased stroke volume | Frank-Starling mechanism |
| Increased afterload | Taller loop; decreased stroke volume; increased ESV | Greater pressure needed to open aortic valve |
| Increased contractility | Steeper ESPVR; decreased ESV; increased stroke volume | Enhanced Ca2+ cycling, catecholamines |
| Heart failure (systolic) | ESPVR shifts right/down; increased ESV; decreased EF | Impaired contractility |
| Aortic stenosis | Higher peak pressure; concentric hypertrophy (steeper EDPVR) | Fixed outflow obstruction |
| Mitral regurgitation | Early decline in LV pressure during systole; increased EDV; decreased effective SV | Low-resistance leak into LA |
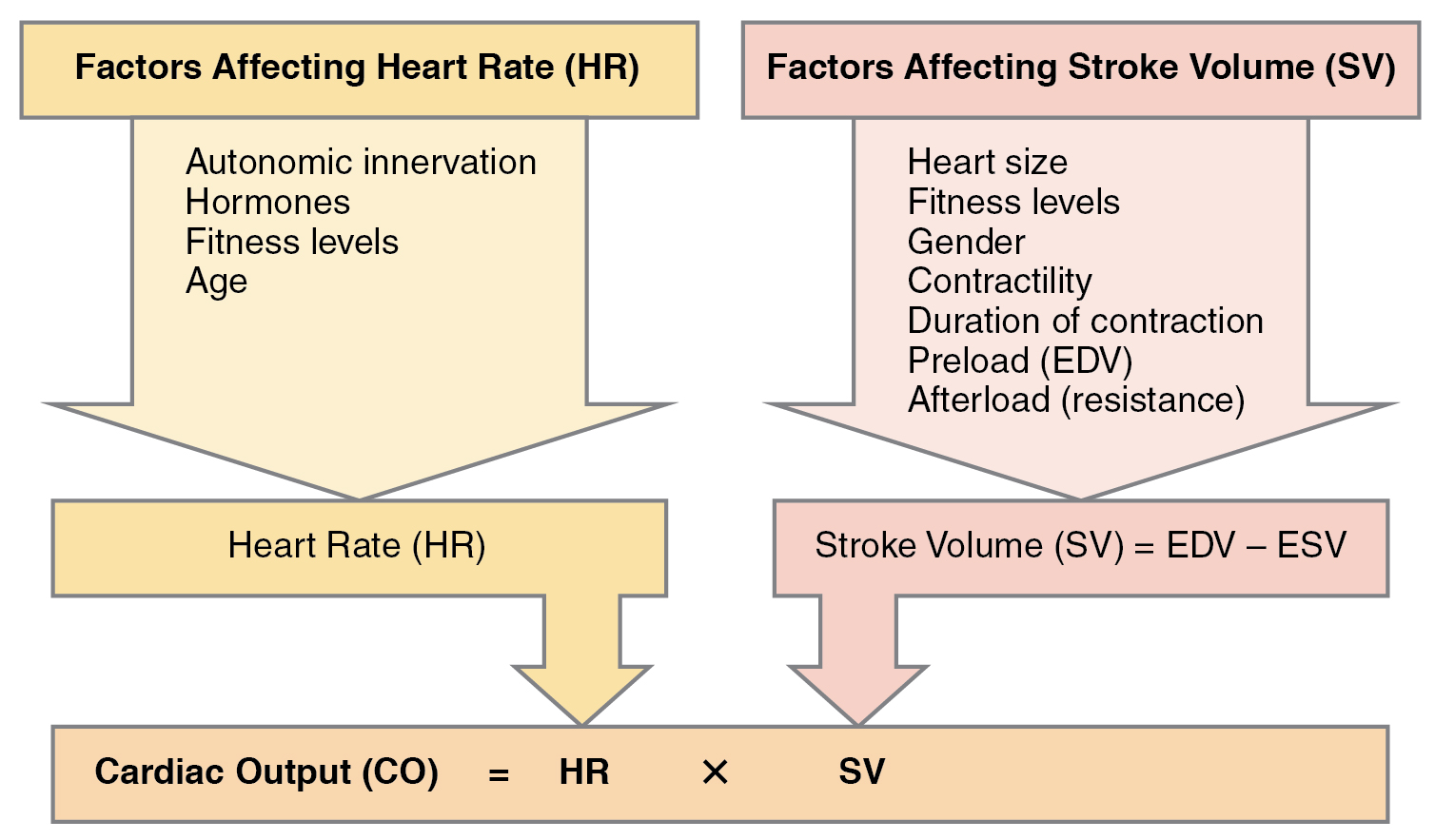
06 Cardiac Output & Hemodynamics
Cardiac Output (CO)
CO = HR x SV (normal ~5 L/min at rest). Stroke volume is determined by three factors: preload (EDV, governed by venous return — Frank-Starling law), afterload (wall stress during ejection, approximated by SVR or aortic pressure), and contractility (intrinsic myocardial force generation independent of loading conditions).
CO = VO2 / (CaO2 - CvO2)
where VO2 = oxygen consumption (~250 mL/min), CaO2 = arterial O2 content, CvO2 = mixed venous O2 content (from PA catheter).
Normal: 250 / (200 - 150) = 250 / 50 = 5 L/min.
This is the gold standard for measuring CO and is frequently tested.
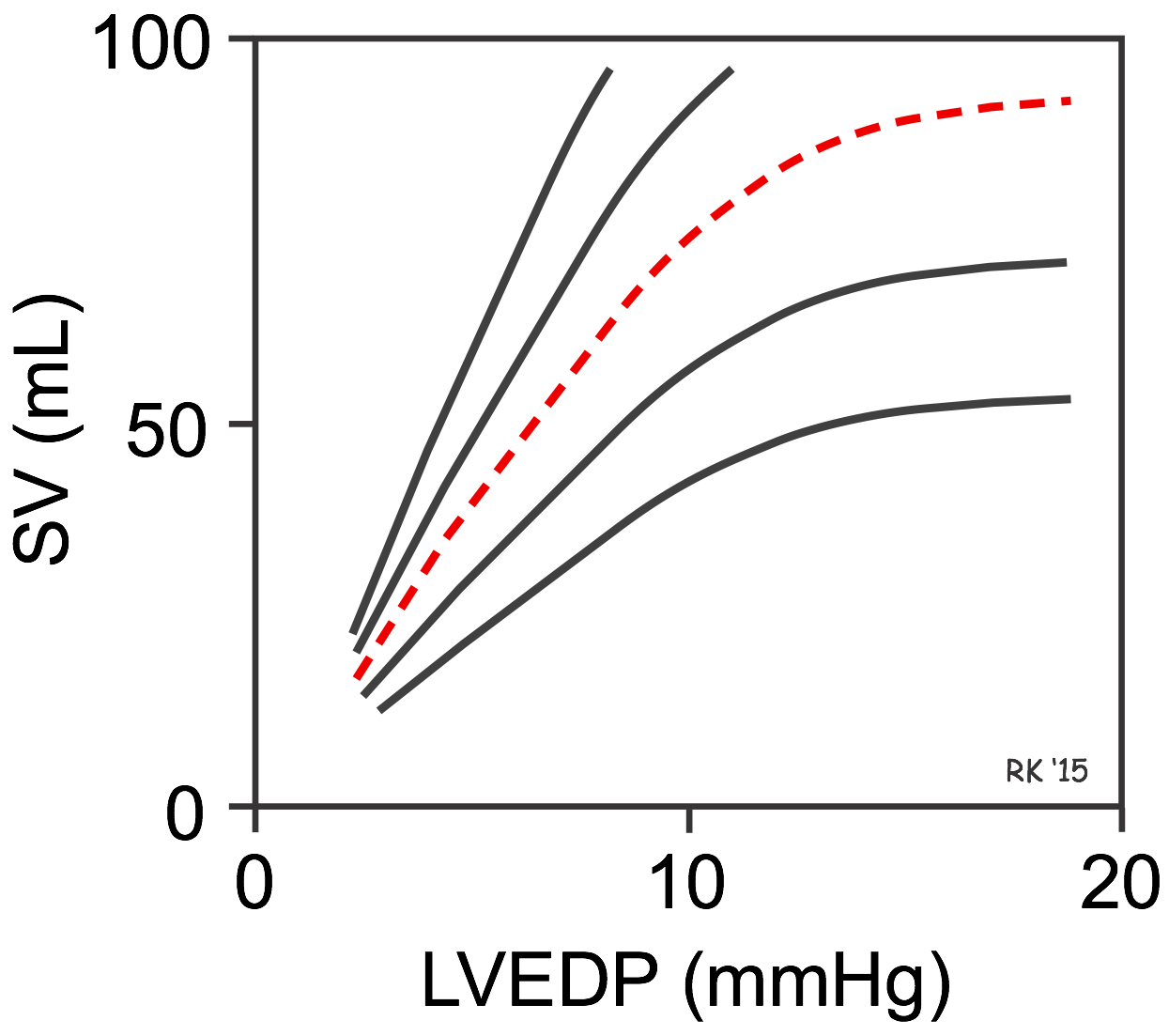
Frank-Starling Law
Within physiologic limits, an increase in venous return (preload, EDV) causes increased sarcomere stretch, which increases the force of contraction and stroke volume. This allows the heart to match its output to venous return beat by beat. The curve shifts up and left with increased contractility (catecholamines, digoxin) and down and right with decreased contractility (heart failure, beta-blockers, acidosis). In severe heart failure, the heart operates on the flat portion of the Starling curve — additional fluid loading does not increase CO but does increase pulmonary congestion.
Mean Arterial Pressure (MAP)
MAP = CO x SVR (the hemodynamic equation). Also approximated as: MAP = DBP + 1/3(SBP - DBP). MAP is the primary determinant of organ perfusion; a MAP below 60-65 mmHg is generally considered inadequate for organ perfusion. MAP is regulated acutely by the baroreceptor reflex and chronically by the RAAS and renal fluid balance.
Ejection Fraction & Cardiac Performance
Ejection fraction (EF) = (SV / EDV) x 100. Normal EF is 55-70%. EF is the most commonly used clinical measure of systolic function. HFrEF (heart failure with reduced ejection fraction, EF < 40%): the ventricle dilates and cannot generate adequate force — systolic failure. HFpEF (heart failure with preserved ejection fraction, EF > 50%): the ventricle is stiff and cannot fill adequately — diastolic failure. EF is load-dependent: it increases with afterload reduction and decreases with mitral regurgitation correction (because the low-resistance "pop-off" into the LA is eliminated). For this reason, EF in MR overestimates true contractile function.
Myocardial Oxygen Demand & Supply
The heart extracts ~70-75% of delivered O2 at rest (highest of any organ) — there is minimal extraction reserve. Therefore, increased myocardial O2 demand can only be met by increasing coronary blood flow. Determinants of myocardial O2 demand: heart rate (most important — tachycardia doubles demand), wall stress (= [pressure x radius] / [2 x wall thickness] — LaPlace law), and contractility. Coronary blood flow occurs predominantly during diastole (the myocardium compresses intramural coronary vessels during systole). This is why tachycardia is particularly harmful — it increases O2 demand while reducing diastolic filling time and thus coronary perfusion. Beta-blockers reduce both heart rate and contractility, decreasing O2 demand and increasing diastolic perfusion time — the physiologic basis for their use in angina and acute MI.
Hemodynamic Profiles
| Condition | CO | SVR | PCWP | CVP |
|---|---|---|---|---|
| Cardiogenic shock | Low | High | High | High |
| Hypovolemic shock | Low | High | Low | Low |
| Septic shock (warm) | High | Low | Low | Low |
| Obstructive (PE/tamponade) | Low | High | High (tamponade) / varies | High |
| Neurogenic shock | Low | Low | Low | Low |
Valvular Heart Disease — Hemodynamic Consequences
| Lesion | Murmur Timing | Hemodynamic Effect | Compensatory Mechanism |
|---|---|---|---|
| Aortic stenosis | Systolic crescendo-decrescendo | Increased LV afterload → pressure overload | Concentric LV hypertrophy (thick walls, normal chamber) |
| Aortic regurgitation | Early diastolic decrescendo | LV volume overload (blood returns during diastole) | Eccentric LV hypertrophy (dilated chamber); wide pulse pressure |
| Mitral stenosis | Low-pitched diastolic rumble with opening snap | LA pressure overload → pulmonary congestion | LA dilation (→ atrial fibrillation risk); pulmonary hypertension |
| Mitral regurgitation | Holosystolic blowing | LV volume overload + LA volume overload | LA and LV dilation; decreased effective forward SV |
07 Vascular Physiology & Blood Pressure Regulation
Vascular Structure & Function
Arteries are high-pressure, elastic vessels that dampen pulsatile flow into steady flow (Windkessel effect). Arterioles are the principal resistance vessels and the major site of blood pressure regulation — small changes in arteriolar radius cause large changes in resistance (Poiseuille's law: resistance is proportional to 1/r4). Capillaries are thin-walled exchange vessels with the largest total cross-sectional area and lowest flow velocity (optimizing diffusion time). Veins are high-capacitance vessels that contain approximately 60-70% of total blood volume and serve as a blood reservoir. Venoconstriction (sympathetic alpha-1) increases venous return and preload.
Starling Forces — Capillary Fluid Exchange
Q = Kf [(Pc - Pi) - sigma(pic - pii)]
Pc = capillary hydrostatic pressure (favors filtration)
Pi = interstitial hydrostatic pressure (opposes filtration)
pic = capillary oncotic pressure (opposes filtration, mainly albumin)
pii = interstitial oncotic pressure (favors filtration)
Kf = filtration coefficient; sigma = reflection coefficient
Edema Formation — Clinical Application of Starling Forces
| Cause of Edema | Starling Force Altered | Clinical Examples |
|---|---|---|
| Increased capillary hydrostatic pressure | Increased Pc | Heart failure (pulmonary and peripheral edema), DVT, venous insufficiency |
| Decreased plasma oncotic pressure | Decreased pic | Nephrotic syndrome, cirrhosis, malnutrition (low albumin) |
| Increased capillary permeability | Increased Kf, decreased sigma | Burns, sepsis, anaphylaxis, ARDS |
| Lymphatic obstruction | Increased pii (protein accumulation) | Filariasis, post-surgical lymphedema, radiation |
Endothelial Function & Vasoactive Mediators
| Mediator | Source | Action | Clinical Significance |
|---|---|---|---|
| Nitric oxide (NO) | Endothelial eNOS (from L-arginine) | Vasodilation (cGMP → smooth muscle relaxation) | Endothelial dysfunction → decreased NO → atherosclerosis; nitroglycerin donates NO |
| Prostacyclin (PGI2) | Endothelium (via COX) | Vasodilation, inhibits platelet aggregation | Balanced against TXA2; epoprostenol for pulmonary hypertension |
| Thromboxane A2 (TXA2) | Platelets (via COX-1) | Vasoconstriction, promotes platelet aggregation | Aspirin irreversibly inhibits COX-1 → decreased TXA2 → antiplatelet effect |
| Endothelin-1 | Endothelium | Potent vasoconstriction | Elevated in pulmonary hypertension; bosentan (ET receptor antagonist) for PAH |
| Bradykinin | Kinin cascade (kallikrein) | Vasodilation, increased vascular permeability | Accumulates with ACE inhibitors → cough, angioedema |
| Histamine | Mast cells | Vasodilation (H1), increased permeability | Anaphylaxis — massive vasodilation and capillary leak |
Blood Pressure Regulation
Autoregulation in Key Vascular Beds
Certain organs maintain constant blood flow over a range of perfusion pressures through intrinsic autoregulatory mechanisms:
| Organ | Autoregulatory Range (MAP) | Key Mediator of Vasodilation | Clinical Significance |
|---|---|---|---|
| Brain | 60-150 mmHg | CO2 (most potent cerebral vasodilator), adenosine, NO | Chronic hypertension shifts curve right → vulnerable to ischemia at "normal" BP |
| Heart | 60-140 mmHg | Adenosine, NO, local metabolites | Coronary flow is highest in diastole (subendocardium vulnerable in aortic stenosis) |
| Kidney | 80-180 mmHg | Myogenic reflex, TGF (adenosine), PGE2 | NSAIDs + ACEi + diuretic = "triple whammy" for AKI |
| Skeletal muscle (resting) | Minimal autoregulation | Local metabolites during exercise | Blood flow increases 20-fold during exercise via metabolic vasodilation |
Acute regulation (seconds-minutes): The baroreceptor reflex uses stretch-sensitive receptors in the carotid sinus (CN IX, glossopharyngeal) and aortic arch (CN X, vagus) to detect changes in MAP. A rise in MAP increases baroreceptor firing, which increases parasympathetic output and decreases sympathetic output — reducing heart rate, contractility, and SVR. A fall in MAP does the opposite. Baroreceptors reset over 1-2 days in chronic hypertension — explaining why rapid lowering of chronically elevated BP can cause syncope.
Intermediate regulation (minutes-hours): The renin-angiotensin system responds to decreased renal perfusion pressure, decreased NaCl delivery to the macula densa, and increased sympathetic stimulation. Angiotensin II is a potent vasoconstrictor that also stimulates aldosterone release and ADH secretion. Atrial natriuretic peptide (ANP) is released from atrial cardiomyocytes in response to stretch, promoting natriuresis and vasodilation.
Chronic regulation (hours-days): The kidneys control blood volume through sodium and water excretion. The pressure-natriuresis mechanism ensures that increased arterial pressure leads to increased Na+ and water excretion, reducing blood volume and returning MAP to normal. This is the ultimate long-term regulator of blood pressure.
Venous Return & Central Venous Pressure
Venous return is driven by the pressure gradient between the mean systemic filling pressure (MSFP, ~7 mmHg — the pressure in the venous system when the heart is stopped) and the right atrial pressure (RAP, ~0-4 mmHg). Venous return = (MSFP - RAP) / venous resistance. Factors that increase venous return: increased blood volume (increases MSFP), venoconstriction (increases MSFP), decreased RAP, muscular compression of veins (muscle pump), negative intrathoracic pressure during inspiration (respiratory pump). The Guyton venous return curve plotted with the Frank-Starling curve demonstrates the equilibrium point of the cardiovascular system: the intersection of the two curves determines the operating CO and RAP. Volume loading shifts the venous return curve right (increased MSFP); heart failure shifts the Starling curve down; positive inotropes shift the Starling curve up.
Microcirculation & Exchange
The capillary bed has the largest total cross-sectional area (~5000 cm2) and the slowest velocity (~0.03 cm/s) of any vascular segment — optimizing time for diffusion. Exchange occurs by three mechanisms: (1) diffusion (the most important — small lipophilic molecules cross freely; small hydrophilic molecules cross through intercellular clefts and fenestrations), (2) bulk flow/filtration (governed by Starling forces), and (3) transcytosis (vesicular transport of large proteins, particularly in the brain — contributing to the blood-brain barrier). Continuous capillaries (most tissues, tight junctions) are the least permeable; fenestrated capillaries (kidney, intestine, endocrine glands) have pores allowing small molecules; sinusoidal capillaries (liver, spleen, bone marrow) have large gaps allowing proteins and even cells to pass.
08 Pulmonary Mechanics & Ventilation
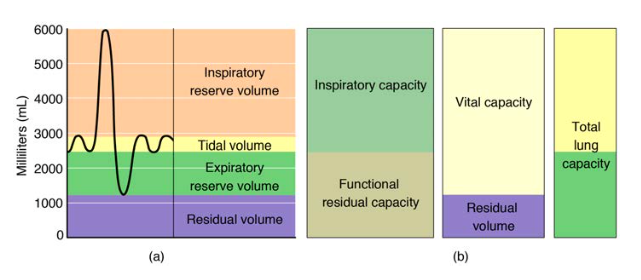
Lung Volumes & Capacities
| Volume/Capacity | Definition | Typical Value (70 kg adult) |
|---|---|---|
| Tidal Volume (TV) | Volume of a normal breath | 500 mL |
| Inspiratory Reserve Volume (IRV) | Extra air that can be inhaled beyond TV | 3000 mL |
| Expiratory Reserve Volume (ERV) | Extra air that can be exhaled beyond TV | 1100 mL |
| Residual Volume (RV) | Air remaining after maximal exhalation | 1200 mL |
| Total Lung Capacity (TLC) | TV + IRV + ERV + RV | 5800 mL |
| Vital Capacity (VC) | TV + IRV + ERV | 4600 mL |
| Functional Residual Capacity (FRC) | ERV + RV | 2300 mL |
| Inspiratory Capacity (IC) | TV + IRV | 3500 mL |
Compliance & Surfactant
Compliance = change in volume / change in pressure (mL/cmH2O). High compliance = easily distensible (emphysema — loss of elastic recoil from alveolar wall destruction). Low compliance = stiff lung (fibrosis, ARDS, pulmonary edema). Surfactant is produced by type II pneumocytes beginning at ~24-28 weeks gestation. It reduces alveolar surface tension, prevents alveolar collapse at end-expiration, and increases compliance. By the Law of Laplace (P = 2T/r), smaller alveoli would have higher collapsing pressure and empty into larger alveoli — surfactant prevents this by reducing surface tension more in smaller alveoli (higher concentration at lower volumes).
Surfactant deficiency causes neonatal respiratory distress syndrome (NRDS) in premature infants. Treatment: exogenous surfactant replacement. Prevention: antenatal corticosteroids (betamethasone) to accelerate surfactant production. The lecithin/sphingomyelin (L/S) ratio in amniotic fluid predicts fetal lung maturity: L/S > 2.0 indicates mature surfactant production; L/S < 1.5 indicates high risk of NRDS.
Pulmonary Pressures & Vascular Physiology
The pulmonary circulation is a low-pressure, low-resistance system. Normal pulmonary arterial pressure is ~25/10 mmHg (mean ~15 mmHg), compared to systemic arterial ~120/80 mmHg (mean ~93 mmHg). Despite receiving the entire cardiac output, the pulmonary circulation operates at ~1/6 the pressure of the systemic circulation because of its low resistance (thin-walled, highly distensible vessels). When pulmonary blood flow increases (e.g., exercise), resistance falls further through recruitment (opening of previously closed capillaries) and distension (widening of already-open capillaries), keeping pressure low. Pulmonary hypertension (mean PAP > 20 mmHg) develops when this compensatory capacity is overwhelmed — causes include left heart disease (most common), lung disease/hypoxia, chronic PE, and primary pulmonary arterial hypertension (PAH). Chronic PH leads to right ventricular hypertrophy and eventually right heart failure (cor pulmonale).
Airway Resistance & Work of Breathing
Resistance = (Palveolar - Patmospheric) / airflow. By Poiseuille's law, resistance is proportional to 1/r4 — halving the radius increases resistance 16-fold. The medium-sized bronchi (generations 4-8) contribute the most to total resistance, not the smallest airways (which have enormous collective cross-sectional area). Factors increasing resistance: bronchospasm, mucus, mucosal edema, airway compression (tumor, dynamic compression during forced expiration). The equal pressure point (EPP) theory explains dynamic airway compression: during forced expiration, pleural pressure exceeds intraluminal pressure at the EPP, causing airway collapse — worsened in emphysema (reduced radial traction) and the basis for pursed-lip breathing (creates back-pressure to stent airways open).
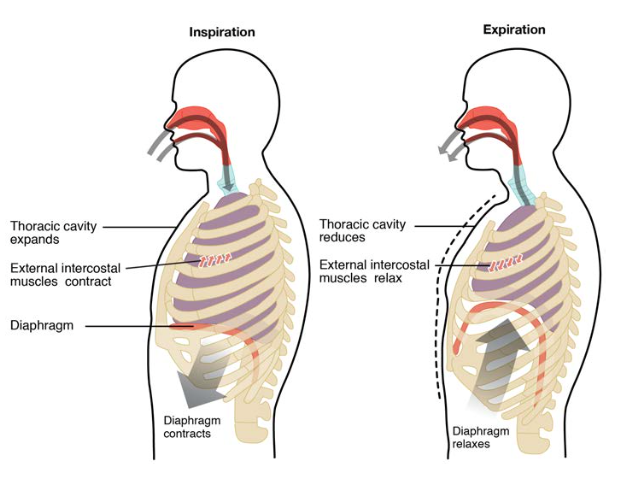
Respiratory Mechanics — Pressure Relationships
Inspiration is normally an active process (diaphragm contraction generates negative intrapleural pressure, ~-5 to -8 cmH2O, which creates a pressure gradient for airflow into the lungs). Expiration is normally passive (elastic recoil of lungs and chest wall). Intrapleural pressure is always negative during quiet breathing (prevents lung collapse). Transpulmonary pressure = alveolar pressure - intrapleural pressure (the distending pressure of the lung; always positive — if it reaches zero, the lung collapses). In pneumothorax, air enters the pleural space → intrapleural pressure becomes atmospheric or positive → transpulmonary pressure falls to zero → lung collapses. In tension pneumothorax, a one-way valve mechanism causes progressive positive intrapleural pressure → mediastinal shift → impaired venous return → cardiovascular collapse. Immediate decompression (needle thoracostomy, 2nd intercostal space, midclavicular line) is life-saving.
Oxygen Cascade — From Atmosphere to Mitochondria
| Location | PO2 (mmHg) | Step |
|---|---|---|
| Atmosphere (dry) | 160 | FiO2 x Patm = 0.21 x 760 |
| Trachea (humidified) | 150 | FiO2 x (Patm - PH2O) = 0.21 x 713 |
| Alveolus (PAO2) | 100 | After CO2 exchange (alveolar gas equation) |
| Arterial blood (PaO2) | 95-100 | Small A-a gradient from physiologic shunt |
| Tissue capillary | 40 | O2 diffuses to tissues along gradient |
| Mitochondria | 1-5 | Minimum PO2 needed for oxidative phosphorylation |
Dead Space & Alveolar Ventilation
Anatomic dead space (~150 mL): conducting airways where no gas exchange occurs. Alveolar dead space: ventilated alveoli with no perfusion (V/Q = infinity). Physiologic dead space = anatomic + alveolar dead space. Alveolar ventilation (VA) = RR x (TV - dead space). Example: RR 12, TV 500 mL: VA = 12 x (500 - 150) = 4200 mL/min. Rapid shallow breathing (e.g., RR 20, TV 250 mL) yields: VA = 20 x (250 - 150) = 2000 mL/min — worse alveolar ventilation despite the same minute ventilation (5000 mL/min), explaining the importance of tidal volume in effective ventilation.
09 Gas Exchange & V/Q Matching
The Alveolar Gas Equation
PAO2 = FiO2 (Patm - PH2O) - (PaCO2 / R)
At sea level on room air: PAO2 = 0.21 (760 - 47) - (40 / 0.8) = 150 - 50 = ~100 mmHg
A-a gradient = PAO2 - PaO2
Normal A-a gradient = 2.5 + (0.21 x age in years)
Young adult: ~5-10 mmHg; Elderly: ~15-20 mmHg
Causes of Hypoxemia
| Mechanism | A-a Gradient | Response to 100% O2 | Classic Examples |
|---|---|---|---|
| Low FiO2 (altitude) | Normal | Corrects | High altitude |
| Hypoventilation | Normal | Corrects | Opioid overdose, obesity hypoventilation, neuromuscular disease |
| V/Q mismatch | Elevated | Corrects | COPD, asthma, pneumonia, PE (most common cause) |
| Diffusion impairment | Elevated | Corrects | Pulmonary fibrosis (exercise-induced desaturation first) |
| Right-to-left shunt | Elevated | Does NOT correct | ARDS, ASD/VSD with Eisenmenger, intrapulmonary shunt |
V/Q Matching & Zones of the Lung
The normal overall V/Q ratio is ~0.8. In the upright lung, gravity creates a gradient: the apex has high V/Q (~3.3, relatively over-ventilated, under-perfused) while the base has low V/Q (~0.6, relatively over-perfused). The West zones describe pulmonary blood flow based on the relationship between PA (alveolar), Pa (arterial), and Pv (venous):
| Zone | Pressure Relationship | Location | Blood Flow |
|---|---|---|---|
| Zone 1 | PA > Pa > Pv | Apex (minimal in health) | No flow — alveolar dead space; appears with hypovolemia or high PEEP |
| Zone 2 | Pa > PA > Pv | Middle lung | Intermittent flow — "waterfall effect"; flow determined by Pa - PA |
| Zone 3 | Pa > Pv > PA | Base | Continuous flow; flow determined by Pa - Pv (classic perfusion) |
10 Oxygen & CO2 Transport
Oxygen Transport
Total O2 content of blood (CaO2) = (1.34 x Hb x SaO2) + (0.003 x PaO2). At normal values: (1.34 x 15 x 0.98) + (0.003 x 100) = 19.7 + 0.3 = 20 mL O2/dL. Dissolved O2 contributes only ~1.5% — virtually all O2 is carried bound to hemoglobin. O2 delivery (DO2) = CO x CaO2 x 10 = 5 x 20 x 10 = ~1000 mL/min. Normal O2 consumption (VO2) ~250 mL/min, so the extraction ratio is ~25% — a large reserve that increases during exercise or low-output states.
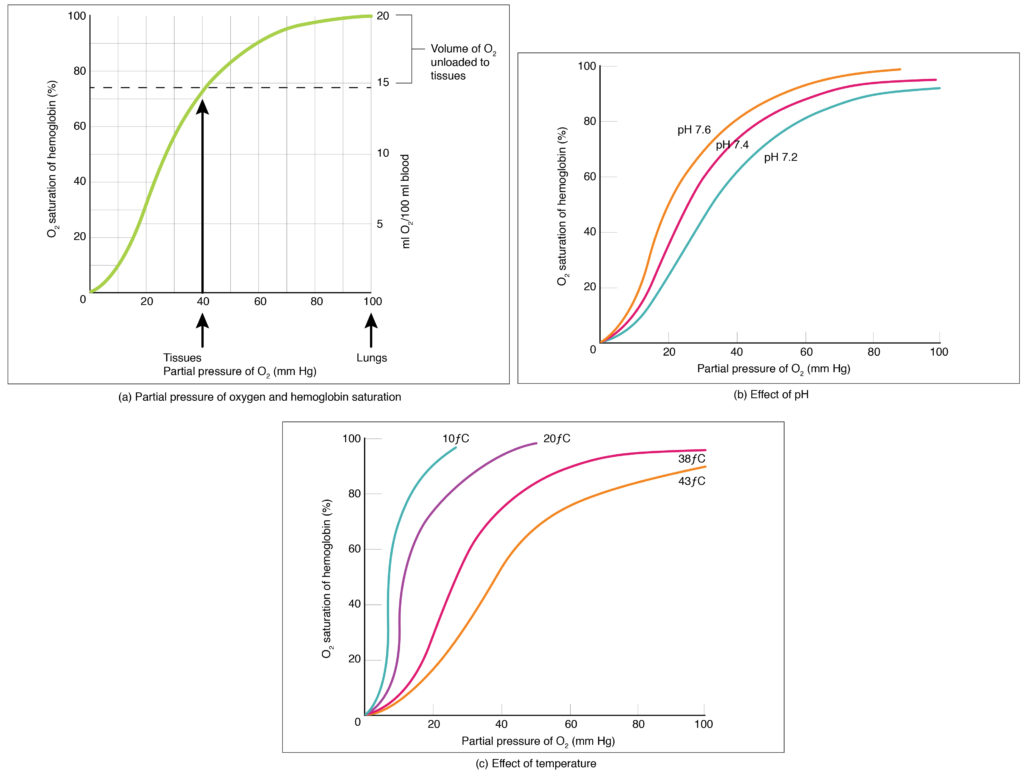
The Oxyhemoglobin Dissociation Curve
The sigmoid shape reflects cooperative binding: once one O2 molecule binds, affinity for subsequent O2 increases (T-state to R-state transition). The P50 is the PO2 at which Hb is 50% saturated — normally ~27 mmHg.
| Right Shift (decreased affinity, more O2 to tissues) | Left Shift (increased affinity, less O2 to tissues) |
|---|---|
| Increased temperature | Decreased temperature (hypothermia) |
| Increased 2,3-DPG | Decreased 2,3-DPG |
| Acidosis / increased CO2 (Bohr effect) | Alkalosis / decreased CO2 |
| High altitude (chronic — via 2,3-DPG) | Carbon monoxide poisoning |
| Exercise (all of the above) | Fetal hemoglobin (HbF) |
| Anemia (compensatory via 2,3-DPG) | Methemoglobinemia |
Chemoreceptor Control of Breathing
Central chemoreceptors (medulla): respond primarily to CSF pH (which reflects PaCO2 — CO2 crosses the blood-brain barrier freely and is converted to H+ + HCO3-). PaCO2 is the strongest drive to breathe under normal conditions. A rise in PaCO2 of just 2-3 mmHg increases ventilation by 100%. Peripheral chemoreceptors: carotid bodies (CN IX → NTS) and aortic bodies (CN X → NTS) respond to decreased PaO2 (primarily, significant response only below ~60 mmHg), increased PaCO2, and decreased pH. The peripheral chemoreceptors are the ONLY sensors that detect hypoxemia.
In chronic CO2 retainers (severe COPD with chronic hypercapnia), central chemoreceptors become desensitized to elevated PaCO2 over time (as CSF HCO3- normalizes pH). These patients rely more heavily on the hypoxic drive via peripheral chemoreceptors. However, the clinical significance of "knocking out the hypoxic drive" with supplemental O2 has been overstated — the dominant mechanism of O2-induced hypercapnia in COPD is actually the Haldane effect (oxygenated Hb releases CO2) and V/Q worsening (reversal of HPV). Regardless, target SpO2 88-92% in acute COPD exacerbation to balance oxygenation against CO2 retention.
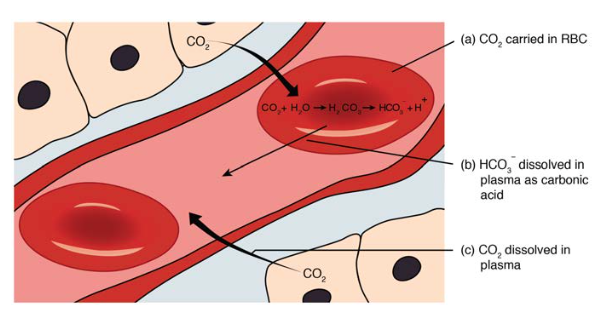
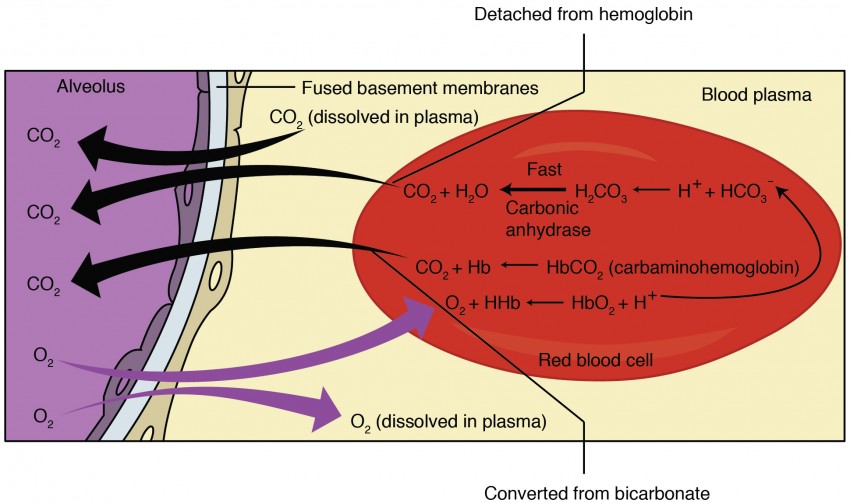
CO2 Transport
CO2 is transported in three forms: dissolved CO2 (~7%), carbaminohemoglobin (~23%, bound to Hb amino groups), and bicarbonate (~70%). In the tissues: CO2 + H2O is converted to H2CO3 (via carbonic anhydrase in RBCs) then to HCO3- + H+. The HCO3- is exchanged for Cl- across the RBC membrane (the chloride shift). In the lungs, this process reverses, releasing CO2 for exhalation. The Haldane effect: deoxygenated Hb carries more CO2 than oxygenated Hb — facilitating CO2 loading in tissues and unloading in the lungs.
11 Glomerular Filtration & Renal Blood Flow
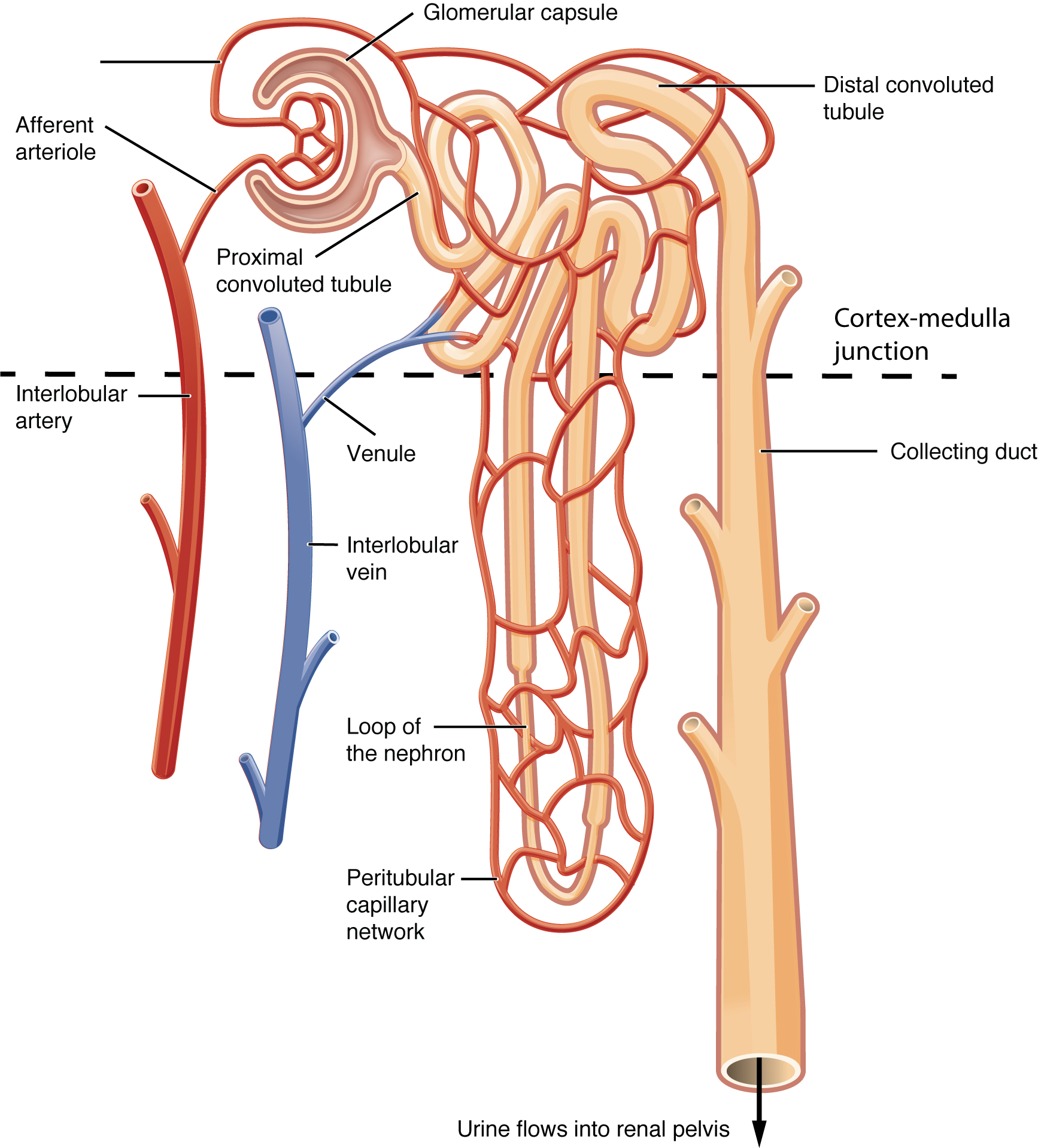
The kidneys receive approximately 25% of cardiac output (~1200 mL/min) despite comprising only 0.4% of body weight — reflecting their critical role in filtration, excretion, and homeostasis. Renal blood flow (RBF) is directed primarily to the cortex (90%), with only 10% to the medulla, which operates in a relatively hypoxic environment (necessary for the concentration gradient).
Glomerular Filtration Rate (GFR)
GFR = Kf x [(PGC - PBS) - (piGC - piBS)]
Normal GFR ~125 mL/min (~180 L/day). Only ~1-2 L/day becomes urine (99% reabsorbed).
Clearance = (Ux x V) / Px
where Ux = urine concentration, V = urine flow rate, Px = plasma concentration.
Inulin clearance = GFR (gold standard — freely filtered, not reabsorbed or secreted).
Creatinine clearance slightly overestimates GFR (some tubular secretion).
PAH clearance = effective renal plasma flow (RPF, ~625 mL/min) — freely filtered AND completely secreted at low concentrations.
Filtration fraction = GFR/RPF = 125/625 = ~20%.
Autoregulation of Renal Blood Flow
RBF and GFR are maintained constant over a MAP range of ~80-180 mmHg through two intrinsic mechanisms: the myogenic mechanism (afferent arteriolar smooth muscle contracts in response to stretch from increased perfusion pressure) and tubuloglomerular feedback (TGF) (increased NaCl delivery to the macula densa causes afferent arteriolar constriction via adenosine, reducing GFR). These mechanisms fail below MAP ~80 mmHg, leading to decreased GFR and prerenal azotemia.
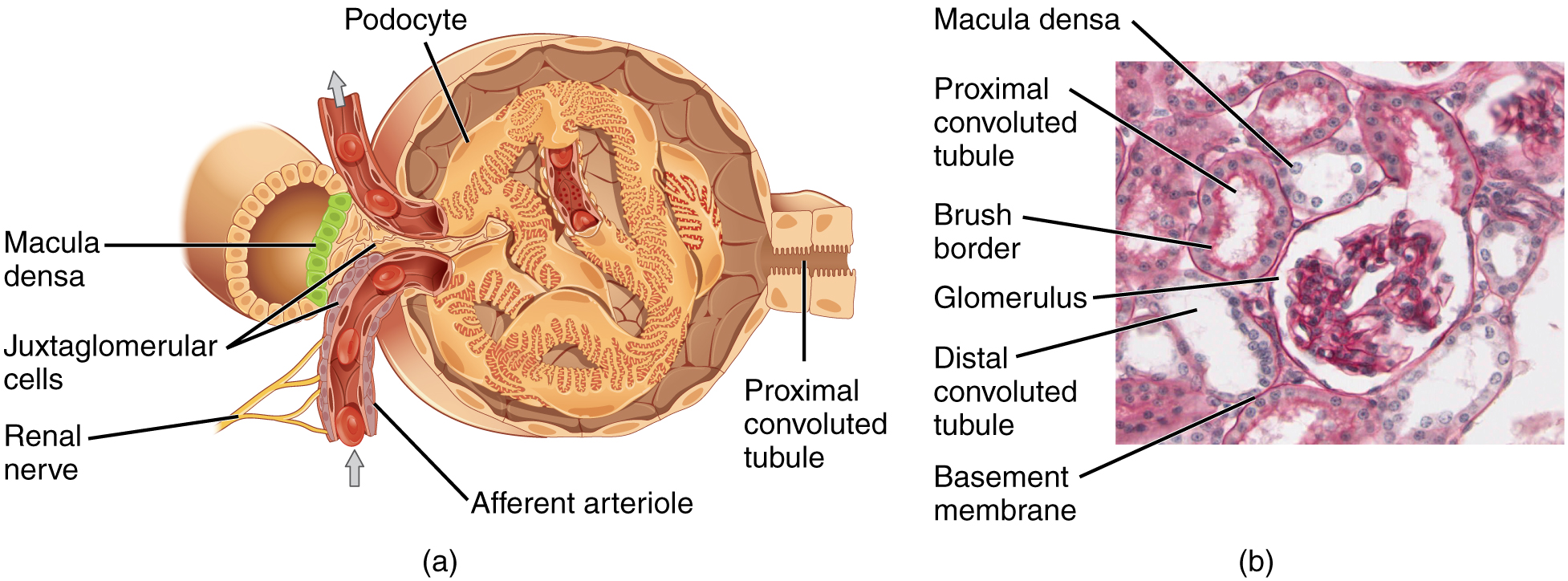
The Juxtaglomerular Apparatus
The JGA consists of three components: (1) Juxtaglomerular (JG) cells — modified smooth muscle cells in the afferent arteriole wall that synthesize, store, and release renin. They act as baroreceptors (decreased stretch → increased renin) and are innervated by sympathetic beta-1 fibers (stimulation → increased renin). (2) Macula densa — specialized epithelial cells in the TAL where it contacts the glomerulus. They sense NaCl concentration in tubular fluid via NKCC2 transport. Decreased NaCl → increased renin release (and afferent dilation via PGE2 → maintains GFR). Increased NaCl → decreased renin and afferent constriction via adenosine. (3) Extraglomerular mesangial cells (lacis cells) — function unclear, may transmit signals between macula densa and JG cells.
Prerenal azotemia: decreased perfusion, intact tubular function. BUN/Cr ratio > 20:1; FENa < 1%; urine osmolality > 500 mOsm/kg; urine Na+ < 20 mEq/L. Tubules appropriately reabsorb Na+ and water to preserve volume.
Intrinsic renal (ATN): tubular damage, impaired reabsorption. BUN/Cr ~10-15:1; FENa > 2%; urine osmolality < 350 mOsm/kg; urine Na+ > 40 mEq/L. Muddy brown granular casts on urinalysis.
FENa = (UNa x PCr) / (PNa x UCr) x 100. Note: FENa is unreliable if the patient is on diuretics — use FEurea instead (< 35% = prerenal).
Regulation of GFR
| Stimulus | Afferent Arteriole | Efferent Arteriole | Effect on GFR | Effect on RPF |
|---|---|---|---|---|
| Angiotensin II (low dose) | Constricts (mild) | Constricts (more) | Maintained/increased | Decreased |
| ACE inhibitors / ARBs | No change | Dilates | Decreased | Increased |
| NSAIDs | Constricts (block PGE2) | No change | Decreased | Decreased |
| Dopamine (low dose) | Dilates | Dilates | Increased | Increased |
Renal Clearance Concepts — Detailed
The clearance of a substance reveals its handling by the nephron relative to GFR:
| Clearance Relationship | Interpretation | Examples |
|---|---|---|
| Clearance = GFR | Freely filtered, no net reabsorption or secretion | Inulin (gold standard), creatinine (approximately) |
| Clearance < GFR | Net reabsorption (substance is reclaimed from tubular fluid) | Glucose (clearance = 0 at normal levels — fully reabsorbed), Na+, amino acids, HCO3-, urea |
| Clearance > GFR | Net secretion (substance is added to tubular fluid beyond filtration) | PAH (at low levels, clearance = RPF), organic acids, penicillin, K+ (in collecting duct) |
| Clearance = 0 | Completely reabsorbed OR not filtered | Glucose at normal levels, albumin (too large to filter) |
The glucose transport maximum (Tm) is ~375 mg/min. At normal plasma glucose (~100 mg/dL) and GFR (125 mL/min), the filtered glucose load is ~125 mg/min — well below Tm, so all glucose is reabsorbed. When plasma glucose exceeds ~180 mg/dL, the filtered load exceeds Tm and glucose "spills" into urine (glycosuria). However, the actual threshold varies between nephrons, creating a "splay" region between ~180-350 mg/dL where some but not all nephrons are at capacity. SGLT2 inhibitors reduce the Tm, producing glycosuria at lower glucose levels.
12 Tubular Transport & Nephron Function
Proximal Convoluted Tubule (PCT)
The workhorse of the nephron — reabsorbs ~65% of filtered Na+, water, K+, HCO3-, glucose, amino acids, phosphate, and urea. All glucose and amino acid reabsorption occurs here via Na+-coupled secondary active transport (SGLT2 for glucose in the early PCT, SGLT1 in the late PCT). The PCT has a high transport maximum (Tm) for glucose (~375 mg/min); when plasma glucose exceeds ~180 mg/dL (renal threshold), glucose appears in the urine (glycosuria). SGLT2 inhibitors (empagliflozin, dapagliflozin) lower the Tm, causing glycosuria at lower glucose levels — the basis for their use in diabetes and heart failure.
Bicarbonate reabsorption in the PCT is mediated by carbonic anhydrase (luminal CA IV and intracellular CA II). The Na+/H+ exchanger (NHE3) on the luminal membrane secretes H+, which combines with filtered HCO3- to form CO2 + H2O (via CA IV). CO2 diffuses into the cell, where CA II regenerates HCO3-, which exits basolaterally. Acetazolamide inhibits CA, causing bicarbonaturia and metabolic acidosis — used for altitude sickness and idiopathic intracranial hypertension.
Loop of Henle
The thin descending limb is permeable to water but not NaCl (concentrates tubular fluid as it descends into the hyperosmolar medulla). The thick ascending limb (TAL) is impermeable to water but actively reabsorbs ~25% of filtered NaCl via the Na+/K+/2Cl- cotransporter (NKCC2). This is the target of loop diuretics (furosemide, bumetanide). The TAL is the "diluting segment" — it makes the tubular fluid hypotonic while making the medullary interstitium hypertonic. The TAL also reabsorbs Mg2+ and Ca2+ paracellularly, driven by the lumen-positive voltage created by K+ recycling through ROMK channels. Loop diuretics abolish this voltage, causing renal Ca2+ and Mg2+ wasting.
Distal Convoluted Tubule (DCT)
Reabsorbs ~5% of filtered NaCl via the Na+/Cl- cotransporter (NCC) — target of thiazide diuretics. The DCT is also a site of active Ca2+ reabsorption (stimulated by PTH and enhanced by thiazides — hence thiazides reduce urinary Ca2+ and are used for recurrent calcium nephrolithiasis).
Collecting Duct
Principal cells: respond to aldosterone (increases ENaC Na+ channels and ROMK K+ channels → Na+ reabsorption and K+ secretion) and ADH (inserts AQP-2 water channels → water reabsorption). Intercalated cells: Type A secrete H+ (acid excretion) and reabsorb HCO3-; Type B secrete HCO3- and reabsorb H+. K+-sparing diuretics act here: spironolactone/eplerenone block mineralocorticoid receptor; amiloride/triamterene directly block ENaC.
Nephron Segment Summary
| Segment | % Na+ Reabsorbed | Key Transporter | Diuretic Target | Water Permeability |
|---|---|---|---|---|
| PCT | 65% | NHE3, SGLT2, Na+/Pi cotransporter | Acetazolamide (CA inhibitor) | High (obligatory with solute) |
| Thin descending limb | 0% | None | None | High (aquaporins) |
| TAL | 25% | NKCC2 | Loop diuretics | Impermeable |
| DCT | 5% | NCC | Thiazides | Impermeable |
| Collecting duct | 3% | ENaC (aldosterone-regulated) | K+-sparing diuretics | ADH-dependent (AQP-2) |
13 Water Balance, ADH & Concentration Mechanisms
Antidiuretic Hormone (ADH / Vasopressin)
ADH is synthesized in the supraoptic and paraventricular nuclei of the hypothalamus, stored in and released from the posterior pituitary. The primary stimulus for ADH release is increased plasma osmolality (detected by osmoreceptors in the hypothalamus; threshold ~280 mOsm/kg). Secondary stimuli include decreased blood volume/pressure (detected by baroreceptors — a less sensitive but more powerful stimulus), pain, nausea, and stress. ADH acts on V2 receptors in the collecting duct, inserting AQP-2 water channels via cAMP-mediated exocytosis, allowing water reabsorption and urine concentration. ADH also acts on V1 receptors on vascular smooth muscle, causing vasoconstriction (hence "vasopressin").
The Countercurrent Multiplier & Exchanger
The medullary osmotic gradient (300 mOsm at the cortex to 1200 mOsm at the papillary tip) is created by the countercurrent multiplier (loop of Henle) and maintained by the countercurrent exchanger (vasa recta). The TAL actively pumps NaCl into the interstitium without water (impermeability), creating a "single effect" of ~200 mOsm at each horizontal level. The countercurrent flow arrangement multiplies this effect along the axis of the medulla. Urea recycling (ADH stimulates urea transporters in the inner medullary collecting duct) contributes ~50% of the medullary osmotic gradient.
Diabetes insipidus (DI): Central DI = insufficient ADH production (treat with desmopressin). Nephrogenic DI = renal resistance to ADH (causes: lithium, hypercalcemia, hypokalemia; treat with thiazides + low-Na+ diet, which paradoxically reduces urine output by causing mild volume contraction and increased proximal reabsorption).
SIADH: Excessive ADH causes water retention, dilutional hyponatremia, concentrated urine (greater than 100 mOsm/kg) with low serum osmolality. Causes: lung cancer (small cell), CNS disorders, drugs (SSRIs, carbamazepine, cyclophosphamide). Treatment: fluid restriction, hypertonic saline if severe; vaptans (tolvaptan) block V2 receptors.
Free Water Clearance
Free water clearance (CH2O) = V - Cosm, where Cosm = (Uosm x V) / Posm. Positive CH2O = dilute urine (water excretion exceeds solute clearance). Negative CH2O (also called TcH2O) = concentrated urine (water is being conserved). In SIADH, CH2O is negative (excessive water retention). In DI, CH2O is positive (excessive water excretion).
Hyponatremia — Physiologic Framework
Hyponatremia (Na+ < 135 mEq/L) is the most common electrolyte disorder. The approach uses serum osmolality, volume status, and urine studies:
| Serum Osmolality | Volume Status | Urine Na+ | Diagnosis |
|---|---|---|---|
| Low (< 280, true hypotonic hyponatremia) | Hypovolemic | < 20 | Extrarenal loss (diarrhea, vomiting, third-spacing) |
| Hypovolemic | > 20 | Renal loss (diuretics, salt-wasting nephropathy, Addison) | |
| Euvolemic | > 20 | SIADH (Uosm > 100), hypothyroidism, adrenal insufficiency | |
| Hypervolemic | Variable | CHF (< 20), cirrhosis (< 20), nephrotic syndrome, renal failure (> 20) | |
| Normal (280-295) | Pseudohyponatremia (hyperlipidemia, hyperproteinemia — lab artifact) | ||
| High (> 295) | Hyperglycemia (Na+ drops ~1.6 mEq/L per 100 mg/dL glucose above normal), mannitol | ||
14 RAAS & Sodium/Potassium Homeostasis
The Renin-Angiotensin-Aldosterone System
Renin is released by juxtaglomerular (JG) cells of the afferent arteriole in response to: (1) decreased renal perfusion pressure, (2) decreased NaCl delivery to the macula densa, (3) increased sympathetic stimulation (beta-1 receptors on JG cells). Renin converts angiotensinogen (from the liver) to angiotensin I. ACE (primarily in the pulmonary vasculature) converts angiotensin I to angiotensin II (Ang II). ACE also degrades bradykinin — ACE inhibitor-induced cough is attributed to bradykinin accumulation.
Actions of Angiotensin II
| Target | Action | Net Effect |
|---|---|---|
| Arterioles (AT1 receptor) | Vasoconstriction (efferent > afferent) | Increased SVR/MAP; maintained GFR |
| Adrenal cortex (zona glomerulosa) | Stimulates aldosterone release | Na+/water retention, K+ excretion |
| Posterior pituitary | Stimulates ADH release | Water retention |
| Hypothalamus | Stimulates thirst | Increased water intake |
| Proximal tubule | Stimulates Na+/HCO3- reabsorption | Volume expansion |
| Sympathetic nervous system | Enhances norepinephrine release | Increased cardiac output and vasoconstriction |
Aldosterone
Released from the zona glomerulosa in response to Ang II, hyperkalemia, and ACTH. It acts on mineralocorticoid receptors in the collecting duct principal cells to increase expression of ENaC (Na+ reabsorption) and ROMK (K+ secretion). Net effect: Na+ retention, K+ excretion, H+ excretion (via increased lumen-negative voltage driving H+ secretion by intercalated cells). Hyperaldosteronism (Conn syndrome): hypertension + hypokalemia + metabolic alkalosis. Hypoaldosteronism (Addison disease, type IV RTA): hypotension + hyperkalemia + metabolic acidosis.
Potassium Homeostasis
98% of total body K+ is intracellular (~140 mEq/L ICF vs ~4 mEq/L ECF). This steep gradient is maintained by the Na+/K+-ATPase and is critical for the resting membrane potential, cardiac conduction, and neuromuscular function. Small changes in serum K+ have profound effects on cardiac electrophysiology — both hypokalemia and hyperkalemia are potentially lethal. Serum K+ is maintained at 3.5-5.0 mEq/L by two mechanisms: internal balance (shifts between ICF and ECF — rapid, minute-to-minute regulation) and external balance (renal excretion, ~90%; GI excretion, ~10% — slower, over hours).
| Shifts K+ INTO Cells (lower serum K+) | Shifts K+ OUT of Cells (raise serum K+) |
|---|---|
| Insulin (stimulates Na+/K+-ATPase) | Insulin deficiency |
| Beta-2 agonists (albuterol) | Beta-blockers |
| Alkalosis | Acidosis (mineral acids > organic acids) |
| Aldosterone | Cell lysis (rhabdomyolysis, tumor lysis, hemolysis) |
| Hypothermia | Hyperosmolality (solvent drag) |
Magnesium & Phosphate
Magnesium (normal 1.5-2.5 mg/dL): 60% in bone, 39% intracellular, 1% ECF. Essential for enzymatic reactions (kinase activity, DNA/RNA synthesis), neuromuscular function, and maintaining K+ and Ca2+ homeostasis. Hypomagnesemia causes refractory hypokalemia (Mg2+ is needed to close ROMK channels in the collecting duct — without it, K+ wasting continues despite supplementation) and refractory hypocalcemia (Mg2+ is needed for PTH secretion and action). Always check and correct Mg2+ when treating hypokalemia or hypocalcemia. Phosphate (normal 2.5-4.5 mg/dL): primarily intracellular, essential for ATP, 2,3-DPG, nucleic acids, bone minerite. Regulated by PTH (phosphaturic) and vitamin D (increases GI absorption). Severe hypophosphatemia (< 1 mg/dL) causes rhabdomyolysis, respiratory failure (diaphragm weakness), hemolytic anemia (decreased RBC ATP and 2,3-DPG), and cardiac dysfunction. Refeeding syndrome in malnourished patients can cause precipitous hypophosphatemia as insulin drives phosphate intracellularly.
15 Acid-Base Fundamentals & Buffer Systems
The Henderson-Hasselbalch Equation
pH = pKa + log([HCO3-] / [0.03 x PaCO2])
pH = 6.1 + log(24 / [0.03 x 40]) = 6.1 + log(24/1.2) = 6.1 + log(20) = 6.1 + 1.3 = 7.40
The ratio of HCO3- to dissolved CO2 must be ~20:1 for normal pH. The kidneys regulate HCO3- (metabolic component); the lungs regulate CO2 (respiratory component).
Buffer Systems
The body uses three buffer systems to resist pH changes: (1) Bicarbonate buffer (CO2/HCO3- system — the most important ECF buffer because both components are independently regulated: CO2 by lungs, HCO3- by kidneys). (2) Hemoglobin buffer (the main ICF buffer in blood — deoxyhemoglobin binds H+ more readily than oxyhemoglobin). (3) Phosphate buffer (HPO42-/H2PO4- — important intracellularly and in renal tubular fluid). Proteins serve as buffers through their amino acid side chains (histidine residues are particularly effective at physiologic pH).
Respiratory Compensation
The lungs respond to metabolic acid-base disturbances within minutes to hours. In metabolic acidosis, peripheral and central chemoreceptors detect low pH, stimulating hyperventilation to lower PaCO2 (Kussmaul breathing in DKA is a classic example). In metabolic alkalosis, hypoventilation raises PaCO2, but this is limited by the hypoxic drive — respiratory compensation for metabolic alkalosis is less effective than for metabolic acidosis.
Renal Compensation
The kidneys respond to respiratory acid-base disturbances over 3-5 days. In respiratory acidosis (elevated PaCO2), the kidneys increase HCO3- reabsorption and H+ excretion (via increased ammoniagenesis and titratable acid). In respiratory alkalosis (low PaCO2), the kidneys decrease HCO3- reabsorption, allowing bicarbonaturia. The renal compensation is slow but powerful and can nearly normalize pH over days.
Renal Acid Excretion
The kidneys excrete approximately 70 mEq of H+ per day (the daily acid load from metabolism). This is accomplished through: (1) HCO3- reabsorption (~4320 mEq/day of filtered HCO3- is reclaimed, primarily in the PCT), (2) titratable acid excretion (~30 mEq/day — H+ buffered by phosphate in tubular fluid: HPO42- + H+ → H2PO4-), and (3) ammonium excretion (~40 mEq/day — NH3 produced in the PCT from glutamine, combines with H+ to form NH4+, which is trapped in the tubular lumen). Ammonium excretion is the primary adaptive response to chronic acid loads and can increase 10-fold in chronic metabolic acidosis.
Urine Anion Gap
Urine anion gap (UAG) = UNa + UK - UCl. This estimates urinary ammonium (NH4+) excretion. In a normal AG metabolic acidosis: a negative UAG (Cl- > Na+ + K+) indicates appropriate renal NH4+ excretion → the acidosis is GI in origin (diarrhea). A positive UAG indicates impaired renal acid excretion → renal tubular acidosis. Remember: "nega-GI-ve" — negative UAG points to GI cause.
16 Acid-Base Disorders & Compensation
Primary Acid-Base Disorders
| Disorder | pH | Primary Change | Compensation | Expected Compensation |
|---|---|---|---|---|
| Metabolic acidosis | Low | Low HCO3- | Decrease PaCO2 | Winter's formula: PaCO2 = 1.5(HCO3-) + 8 +/- 2 |
| Metabolic alkalosis | High | High HCO3- | Increase PaCO2 | PaCO2 = 0.7(HCO3-) + 21 +/- 2 |
| Respiratory acidosis (acute) | Low | High PaCO2 | Increase HCO3- | HCO3- rises 1 mEq/L per 10 mmHg rise in PaCO2 |
| Respiratory acidosis (chronic) | Low-normal | High PaCO2 | Increase HCO3- | HCO3- rises 3.5 mEq/L per 10 mmHg rise in PaCO2 |
| Respiratory alkalosis (acute) | High | Low PaCO2 | Decrease HCO3- | HCO3- falls 2 mEq/L per 10 mmHg fall in PaCO2 |
| Respiratory alkalosis (chronic) | High-normal | Low PaCO2 | Decrease HCO3- | HCO3- falls 5 mEq/L per 10 mmHg fall in PaCO2 |
Anion Gap
Anion gap (AG) = Na+ - (Cl- + HCO3-). Normal = 12 +/- 2 mEq/L (or 8-10 with modern assays). An elevated AG indicates the presence of unmeasured anions — the mnemonic MUDPILES: Methanol, Uremia, DKA, Propylene glycol, INH/Iron, Lactic acidosis, Ethylene glycol, Salicylates. A normal AG (hyperchloremic) metabolic acidosis results from HCO3- loss (diarrhea, RTA types 1, 2, and 4) or failure to excrete H+.
Delta-Delta Analysis
The delta-delta (delta ratio) compares the change in AG to the change in HCO3-: Delta ratio = (AG - 12) / (24 - HCO3-). If ratio is less than 1: concurrent non-AG metabolic acidosis (HCO3- fell more than expected from the AG elevation alone). If ratio is greater than 2: concurrent metabolic alkalosis (HCO3- did not fall as much as expected). If ratio is 1-2: pure AG metabolic acidosis.
Step 1: Look at pH — acidemia or alkalemia?
Step 2: Identify the primary disorder — does PaCO2 or HCO3- explain the pH?
Step 3: Calculate expected compensation — is compensation appropriate?
Step 4: If metabolic acidosis: calculate anion gap.
Step 5: If elevated AG: calculate delta-delta to check for additional disorders.
This systematic approach identifies simple disorders and mixed acid-base disturbances.
Renal Tubular Acidosis (RTA)
| Feature | Type I (Distal) | Type II (Proximal) | Type IV (Hypoaldosteronism) |
|---|---|---|---|
| Defect | Cannot secrete H+ in collecting duct | Cannot reabsorb HCO3- in PCT | Aldosterone deficiency or resistance |
| Serum K+ | Low (hypokalemia) | Low (hypokalemia) | High (hyperkalemia) |
| Urine pH | > 5.5 (always) | > 5.5 initially, then < 5.5 | < 5.5 |
| Serum HCO3- | Can be very low (< 10) | Moderately low (12-20) | Mildly low (15-20) |
| Complications | Nephrocalcinosis, kidney stones | Osteomalacia (phosphate wasting) | Hyperkalemia |
| Causes | Sjogren, SLE, amphotericin B, lithium | Fanconi syndrome, carbonic anhydrase inhibitors, myeloma | Diabetic nephropathy, ACEi/ARBs, K+-sparing diuretics, Addison |
Osmolar Gap
Osmolar gap = measured osmolality - calculated osmolality. Calculated osmolality = 2(Na+) + glucose/18 + BUN/2.8. Normal osmolar gap is less than 10 mOsm/kg. An elevated osmolar gap with an elevated anion gap metabolic acidosis strongly suggests toxic alcohol ingestion: methanol (→ formic acid, causes blindness), ethylene glycol (→ oxalic acid, causes renal failure with calcium oxalate crystals), or isopropyl alcohol (elevated osmolar gap WITHOUT acidosis — metabolized to acetone, not an acid).
17 GI Motility & Secretion
GI Motility
The GI tract has intrinsic motility generated by the interstitial cells of Cajal (ICC) — the pacemaker cells that generate slow waves (basic electrical rhythm). Slow wave frequency varies by location: stomach ~3/min, duodenum ~12/min, ileum ~8/min. Slow waves set the maximum frequency of contraction, but contraction only occurs when slow waves are depolarized above threshold by excitatory stimuli (acetylcholine, substance P). The migrating motor complex (MMC) occurs during fasting, sweeping undigested material and bacteria from the small intestine in ~90-minute cycles. It is mediated by motilin and disrupted by feeding. Erythromycin acts as a motilin agonist, promoting gastric emptying.
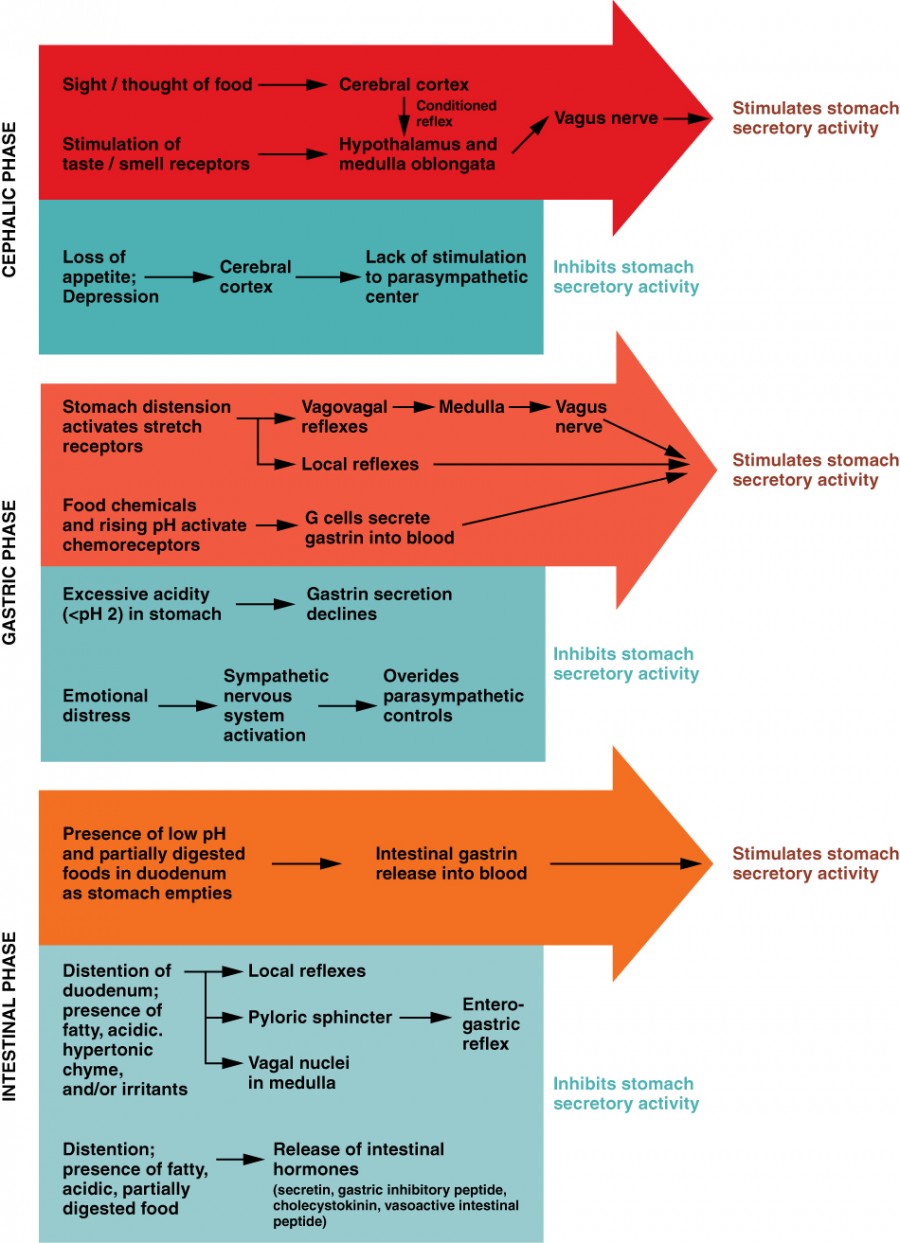
Gastric Acid Secretion
Parietal cells secrete HCl via the H+/K+-ATPase (proton pump) on the luminal surface. Three stimulatory pathways converge on the parietal cell: (1) Acetylcholine (vagal, via M3 receptors — blocked by atropine), (2) Histamine (from ECL cells, via H2 receptors — blocked by H2 blockers such as famotidine), (3) Gastrin (from G cells, via CCK-B receptors). PPIs (omeprazole, pantoprazole) irreversibly inhibit the H+/K+-ATPase — the most potent acid-suppressing agents.
| Phase | Stimulus | Mediator | % of Total Acid Secretion |
|---|---|---|---|
| Cephalic | Sight, smell, taste, thought of food | Vagus nerve → ACh | 30% |
| Gastric | Gastric distension, peptides | Gastrin (G cells), local reflexes | 60% |
| Intestinal | Amino acids in duodenum | Intestinal gastrin | 10% |
Acid secretion is inhibited by: somatostatin (from D cells — inhibits gastrin, histamine, and acid directly), secretin (from S cells, released by acid in the duodenum), GIP, and prostaglandins (PGE2 — protective; NSAIDs block PGE2 synthesis, promoting ulceration).
Pancreatic & Biliary Secretion
Secretin (released by duodenal S cells in response to acid) stimulates pancreatic ductal cells to secrete HCO3--rich fluid, neutralizing gastric acid. CCK (released by duodenal I cells in response to fatty acids and amino acids) stimulates pancreatic acinar cells to secrete digestive enzymes (trypsinogen, lipase, amylase) and causes gallbladder contraction and sphincter of Oddi relaxation. Bile is produced by hepatocytes, stored and concentrated in the gallbladder, and released into the duodenum for fat emulsification. Bile salts are recycled via the enterohepatic circulation (95% reabsorbed in the terminal ileum via ASBT transporter — this is why terminal ileum resection causes bile salt malabsorption, fat malabsorption, and steatorrhea).
18 Digestion, Absorption & Hepatobiliary Function
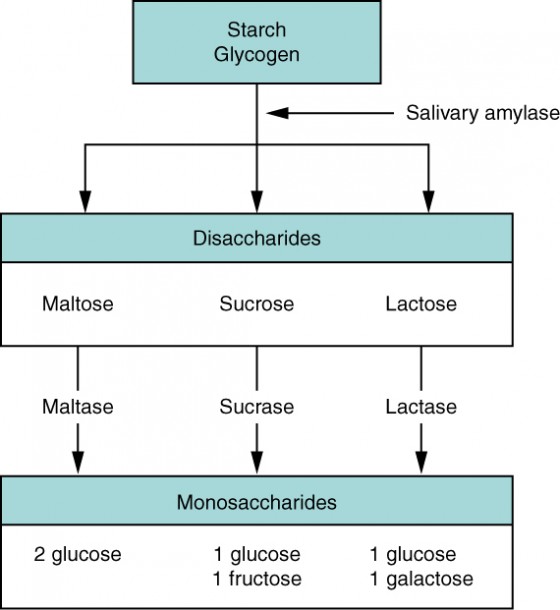
Carbohydrate Digestion & Absorption
Salivary amylase begins starch digestion (inactivated by gastric acid). Pancreatic amylase continues digestion to oligosaccharides in the duodenum. Brush border enzymes (maltase, sucrase, isomaltase, lactase) cleave oligosaccharides and disaccharides into monosaccharides. Glucose and galactose are absorbed via SGLT1 (Na+-coupled active transport) on the luminal surface and exit basolaterally via GLUT2. Fructose is absorbed via GLUT5 (facilitated diffusion). Lactase deficiency is the most common disaccharidase deficiency — undigested lactose is fermented by colonic bacteria, producing gas, bloating, and osmotic diarrhea.
Protein Digestion & Absorption
Pepsin (from pepsinogen, activated by gastric acid) begins protein digestion in the stomach. Pancreatic proteases (trypsin, chymotrypsin, elastase, carboxypeptidases) continue digestion in the duodenum. Enterokinase (brush border enzyme in the duodenum) converts trypsinogen to trypsin, which then activates all other pancreatic zymogens — the key regulatory step. Amino acids are absorbed via Na+-coupled transporters. Di- and tripeptides are absorbed via the PepT1 H+-coupled cotransporter.
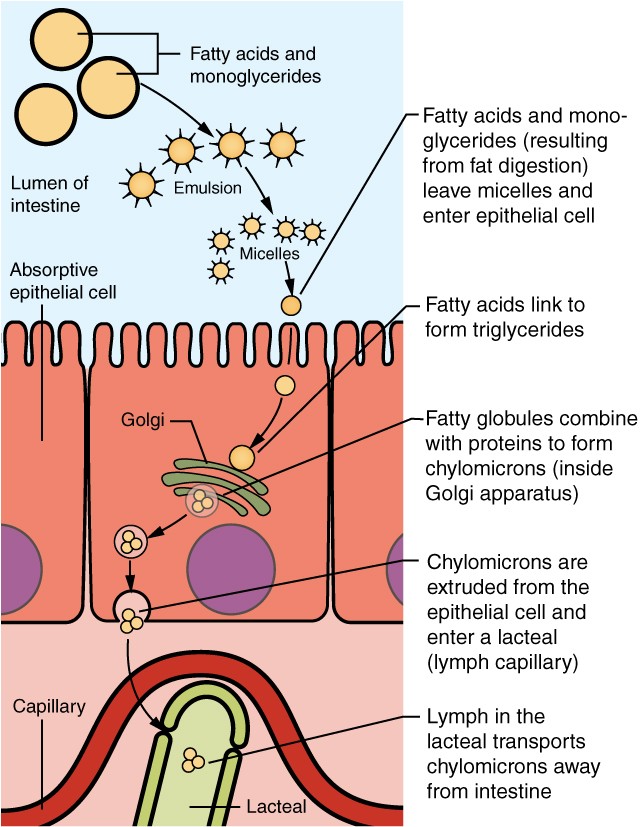
Fat Digestion & Absorption
Lingual and gastric lipases begin triglyceride hydrolysis. Pancreatic lipase (with colipase) is the principal enzyme, cleaving triglycerides at the sn-1 and sn-3 positions to yield 2-monoacylglycerol and free fatty acids. These are incorporated into mixed micelles (with bile salts, cholesterol, and fat-soluble vitamins) for transport across the unstirred water layer. At the brush border, micelle components are absorbed; bile salts remain in the lumen for reabsorption in the terminal ileum. Inside enterocytes, long-chain fatty acids are re-esterified to triglycerides and packaged into chylomicrons, which enter the lymphatics (lacteals) and eventually the bloodstream via the thoracic duct. Short- and medium-chain fatty acids enter the portal blood directly.
Intestinal Fluid & Electrolyte Transport
The GI tract handles approximately 9 L of fluid daily (2 L ingested + 7 L of secretions from salivary glands, stomach, pancreas, bile, and intestine). The small intestine reabsorbs ~7.5 L, the colon ~1.3 L, leaving only ~100-200 mL in stool. Disruption of this balance causes diarrhea. Secretory diarrhea: active secretion of Cl-/water into the lumen (cholera toxin → cAMP → CFTR Cl- channel activation; large volume, persists during fasting, no osmotic gap). Osmotic diarrhea: unabsorbed solutes draw water into the lumen (lactose intolerance, sorbitol, magnesium-containing antacids; stops during fasting, positive osmotic gap: stool osmolality - 2[stool Na+ + stool K+] > 50 mOsm). Oral rehydration solution (ORS) exploits the Na+/glucose cotransporter (SGLT1) — glucose facilitates Na+ absorption, and water follows osmotically. This is why ORS contains both glucose and sodium and has saved millions of lives from cholera and infectious diarrhea worldwide.
Absorption Sites
| Nutrient | Primary Absorption Site | Clinical Significance of Disease at That Site |
|---|---|---|
| Iron | Duodenum | Celiac disease → iron deficiency anemia |
| Folate | Jejunum | Celiac disease, tropical sprue → megaloblastic anemia |
| Vitamin B12 | Terminal ileum | Crohn disease (terminal ileum), pernicious anemia (no IF) → megaloblastic anemia |
| Bile salts | Terminal ileum | Ileal resection → bile salt wasting → fat malabsorption/steatorrhea |
| Fat-soluble vitamins (A, D, E, K) | Proximal small intestine (with fat) | Pancreatic insufficiency, bile salt deficiency → deficiency states |
| Calcium | Duodenum (active, vitamin D-dependent) | Vitamin D deficiency, celiac disease → hypocalcemia |
| Water & electrolytes | Small intestine (majority) & colon | Cholera toxin (cAMP) → secretory diarrhea |
Vitamin Absorption & Deficiency Syndromes
| Vitamin | Absorption Mechanism | Deficiency Consequences | At-Risk Populations |
|---|---|---|---|
| A (retinol) | Fat-soluble; absorbed with micelles in proximal SI | Night blindness, xerophthalmia, Bitot spots, immune dysfunction | Fat malabsorption, CF, cholestasis |
| D (cholecalciferol) | Fat-soluble; also synthesized in skin (UV-B) | Rickets (children), osteomalacia (adults), hypocalcemia | Limited sun exposure, CKD, malabsorption |
| E (tocopherol) | Fat-soluble; absorbed with micelles | Hemolytic anemia, posterior column/spinocerebellar degeneration (mimics B12 neuro but no megaloblastic anemia) | Abetalipoproteinemia, CF, cholestasis |
| K (phylloquinone) | Fat-soluble; also produced by gut bacteria | Bleeding diathesis (decreased factors II, VII, IX, X, protein C & S) | Newborns (given vitamin K at birth), warfarin use, bile duct obstruction |
| B1 (thiamine) | Active transport in jejunum | Wernicke encephalopathy (confusion, ataxia, ophthalmoplegia), beriberi (wet = cardiac, dry = neuro) | Alcoholism, malnutrition; always give BEFORE glucose in malnourished patients |
| B12 (cobalamin) | Requires intrinsic factor (from parietal cells); absorbed in terminal ileum | Megaloblastic anemia + neurologic symptoms (subacute combined degeneration: posterior columns + lateral corticospinal tracts) | Pernicious anemia, vegan diet, Crohn (terminal ileum), metformin use |
| Folate (B9) | Absorbed in jejunum (no IF needed) | Megaloblastic anemia WITHOUT neurologic symptoms; neural tube defects in pregnancy | Alcoholism, pregnancy, methotrexate/trimethoprim use (DHFR inhibitors) |
Hepatic Function
The liver performs over 500 functions including: bile synthesis, bilirubin conjugation and excretion, albumin synthesis (major determinant of plasma oncotic pressure), coagulation factor synthesis (I, II, V, VII, IX, X, XI, XII, XIII), drug metabolism (phase I: CYP450 oxidation; phase II: conjugation), ammonia metabolism (urea cycle), glycogen storage and gluconeogenesis, cholesterol and lipoprotein metabolism, and detoxification. Hepatic blood flow (~1500 mL/min) arrives via the portal vein (~75%, deoxygenated, nutrient-rich) and the hepatic artery (~25%, oxygenated).
Bilirubin Metabolism
Bilirubin is the breakdown product of heme (from senescent RBCs, ~80%). The pathway: heme → biliverdin (by heme oxygenase) → unconjugated (indirect) bilirubin (water-insoluble, albumin-bound in blood, NOT filtered by kidneys). In hepatocytes: unconjugated bilirubin is conjugated with glucuronic acid by UDP-glucuronyl transferase (UGT1A1) → conjugated (direct) bilirubin (water-soluble) → excreted into bile → intestinal bacteria convert it to urobilinogen → most excreted in stool as stercobilin (brown color); ~10-20% reabsorbed (enterohepatic circulation) → some excreted by kidney as urobilin (yellow urine color).
| Condition | Unconjugated (Indirect) | Conjugated (Direct) | Urine Bilirubin | Stool Color |
|---|---|---|---|---|
| Hemolysis | Elevated | Normal | Absent | Dark (increased stercobilin) |
| Gilbert syndrome | Mildly elevated | Normal | Absent | Normal |
| Hepatocellular damage | Elevated | Elevated | Present | Normal to pale |
| Obstructive jaundice | Normal | Elevated | Present (dark urine) | Pale/clay-colored (acholic) |
GI Hormones Summary
| Hormone | Source | Stimulus | Actions |
|---|---|---|---|
| Gastrin | G cells (antrum) | Peptides, vagal stimulation, gastric distension | Stimulates HCl secretion, gastric mucosal growth |
| CCK | I cells (duodenum/jejunum) | Fatty acids, amino acids | Gallbladder contraction, pancreatic enzyme secretion, sphincter of Oddi relaxation, satiety |
| Secretin | S cells (duodenum) | Acid in duodenum | Pancreatic HCO3- secretion, inhibits gastrin |
| GIP | K cells (duodenum/jejunum) | Glucose, fatty acids | Stimulates insulin release (incretin effect), inhibits gastric acid |
| GLP-1 | L cells (ileum/colon) | Glucose, fatty acids | Stimulates insulin, inhibits glucagon, delays gastric emptying, satiety |
| Motilin | M cells (duodenum/jejunum) | Fasting state | Initiates MMC; erythromycin = motilin agonist |
| VIP | Parasympathetic neurons | Neural stimulation | Relaxes smooth muscle, stimulates pancreatic/intestinal secretion; VIPoma → watery diarrhea |
| Somatostatin | D cells (throughout GI) | Acid, fat | Inhibits virtually all GI hormones and secretions ("universal inhibitor") |
19 Neuronal Signaling & Synaptic Transmission
The Action Potential
Neurons communicate via action potentials — all-or-none electrical events that propagate along axons. At rest, the membrane is at approximately -70 mV (K+ leak channels dominate). When a stimulus depolarizes the membrane to threshold (~-55 mV), voltage-gated Na+ channels open rapidly, causing a positive feedback loop of depolarization (phase 0, rising to ~+30 mV). Na+ channels then inactivate (refractory period) while voltage-gated K+ channels open, repolarizing the membrane (phase 3). A brief hyperpolarization (undershoot) occurs as K+ channels close slowly. The absolute refractory period (Na+ channels inactivated — no AP possible regardless of stimulus) ensures unidirectional propagation. The relative refractory period (some Na+ channels recovered — AP possible with stronger-than-normal stimulus) allows frequency coding of stimulus intensity.
Conduction Velocity
AP propagation speed depends on axon diameter (larger = faster) and myelination. Myelinated axons conduct via saltatory conduction — APs "jump" between nodes of Ranvier, where Na+ channels are concentrated. This increases velocity from ~1 m/s (unmyelinated C fibers, pain) to ~120 m/s (large myelinated A-alpha fibers, proprioception). Demyelinating diseases (multiple sclerosis, Guillain-Barre) slow or block conduction, causing neurologic deficits.
Nerve Fiber Classification
| Fiber Type | Diameter | Velocity | Myelination | Function |
|---|---|---|---|---|
| A-alpha | 12-20 micrometers | 70-120 m/s | Heavy | Proprioception, motor (alpha motor neurons) |
| A-beta | 5-12 micrometers | 30-70 m/s | Heavy | Touch, pressure |
| A-gamma | 3-6 micrometers | 15-30 m/s | Moderate | Muscle spindle motor (gamma motor neurons) |
| A-delta | 2-5 micrometers | 12-30 m/s | Light | Fast (sharp, well-localized) pain, temperature, touch |
| B | < 3 micrometers | 3-15 m/s | Light | Preganglionic autonomic |
| C | 0.4-1.2 micrometers | 0.5-2 m/s | Unmyelinated | Slow (dull, diffuse) pain, temperature; postganglionic autonomic |
Synaptic Transmission
At chemical synapses: AP arrival at the presynaptic terminal opens voltage-gated Ca2+ channels → Ca2+ influx triggers vesicle fusion (SNARE complex: synaptobrevin, SNAP-25, syntaxin) → neurotransmitter release into the synaptic cleft → binding to postsynaptic receptors. Neurotransmitter is then removed by reuptake (monoamine transporters — target of SSRIs, SNRIs, TCAs, cocaine), enzymatic degradation (AChE breaks down ACh; MAO degrades monoamines), or diffusion.
Major Neurotransmitters
| Neurotransmitter | Receptor Types | Function | Clinical Relevance |
|---|---|---|---|
| Acetylcholine (ACh) | Nicotinic (ionotropic), Muscarinic (M1-M5, GPCR) | NMJ, parasympathetic, memory | Myasthenia gravis (anti-AChR Ab); cholinesterase inhibitors for Alzheimer |
| Norepinephrine | Alpha-1,2; Beta-1,2,3 (GPCR) | Sympathetic, alertness, mood | Depression (target of SNRIs); pheochromocytoma (excess) |
| Dopamine | D1-D5 (GPCR) | Reward, motor, prolactin inhibition | Parkinson (D2 agonists); schizophrenia (D2 antagonists) |
| Serotonin (5-HT) | 5-HT1-7 (mostly GPCR; 5-HT3 ionotropic) | Mood, sleep, GI motility | Depression (SSRIs); carcinoid (excess 5-HT); migraine (triptans = 5-HT1B/1D agonists) |
| GABA | GABA-A (Cl- channel), GABA-B (GPCR) | Major inhibitory NT in CNS | Benzodiazepines (enhance GABA-A); barbiturates; hepatic encephalopathy (excess GABA tone) |
| Glutamate | NMDA, AMPA, Kainate (ionotropic); mGluR (GPCR) | Major excitatory NT in CNS | Excitotoxicity in stroke; ketamine (NMDA antagonist); memantine for Alzheimer |
| Glycine | Glycine receptor (Cl- channel) | Inhibitory in spinal cord/brainstem | Strychnine poisoning (glycine antagonist → convulsions) |
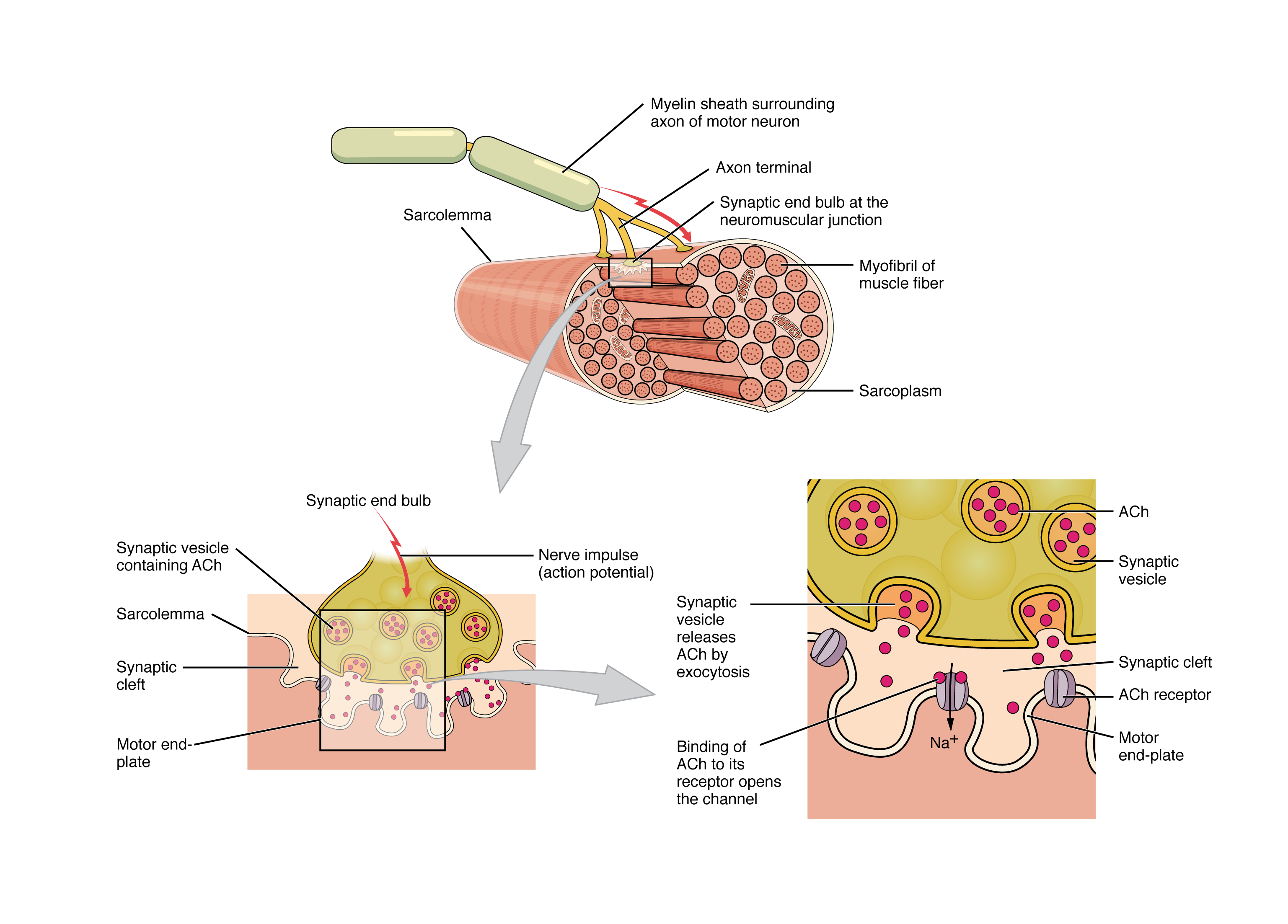
Neuromuscular Junction Physiology
The NMJ is the synapse between a motor neuron and skeletal muscle. An AP arriving at the motor nerve terminal opens voltage-gated Ca2+ channels → Ca2+ influx triggers ACh vesicle release → ACh crosses the synaptic cleft and binds nicotinic ACh receptors (ligand-gated Na+/K+ channels) on the motor end plate → end-plate potential → if threshold is reached, a muscle fiber AP is generated → contraction. ACh is rapidly hydrolyzed by acetylcholinesterase (AChE) in the synaptic cleft, terminating the signal.
| NMJ Disorder | Pathology | Key Features | Treatment |
|---|---|---|---|
| Myasthenia gravis | Antibodies against nicotinic AChR (85%) or MuSK (5-10%) | Fatigable weakness (worse with use), ptosis, diplopia; improves with AChE inhibitors | Pyridostigmine, immunosuppression, thymectomy |
| Lambert-Eaton | Antibodies against presynaptic voltage-gated Ca2+ channels | Proximal weakness that IMPROVES with repeated use; associated with SCLC | Treat underlying cancer, 3,4-DAP, IVIG |
| Organophosphate toxicity | Irreversible AChE inhibition → ACh accumulation | DUMBELS: Diarrhea, Urination, Miosis, Bradycardia, Emesis, Lacrimation, Salivation | Atropine (muscarinic blocker) + pralidoxime (reactivates AChE) |
20 Autonomic Nervous System
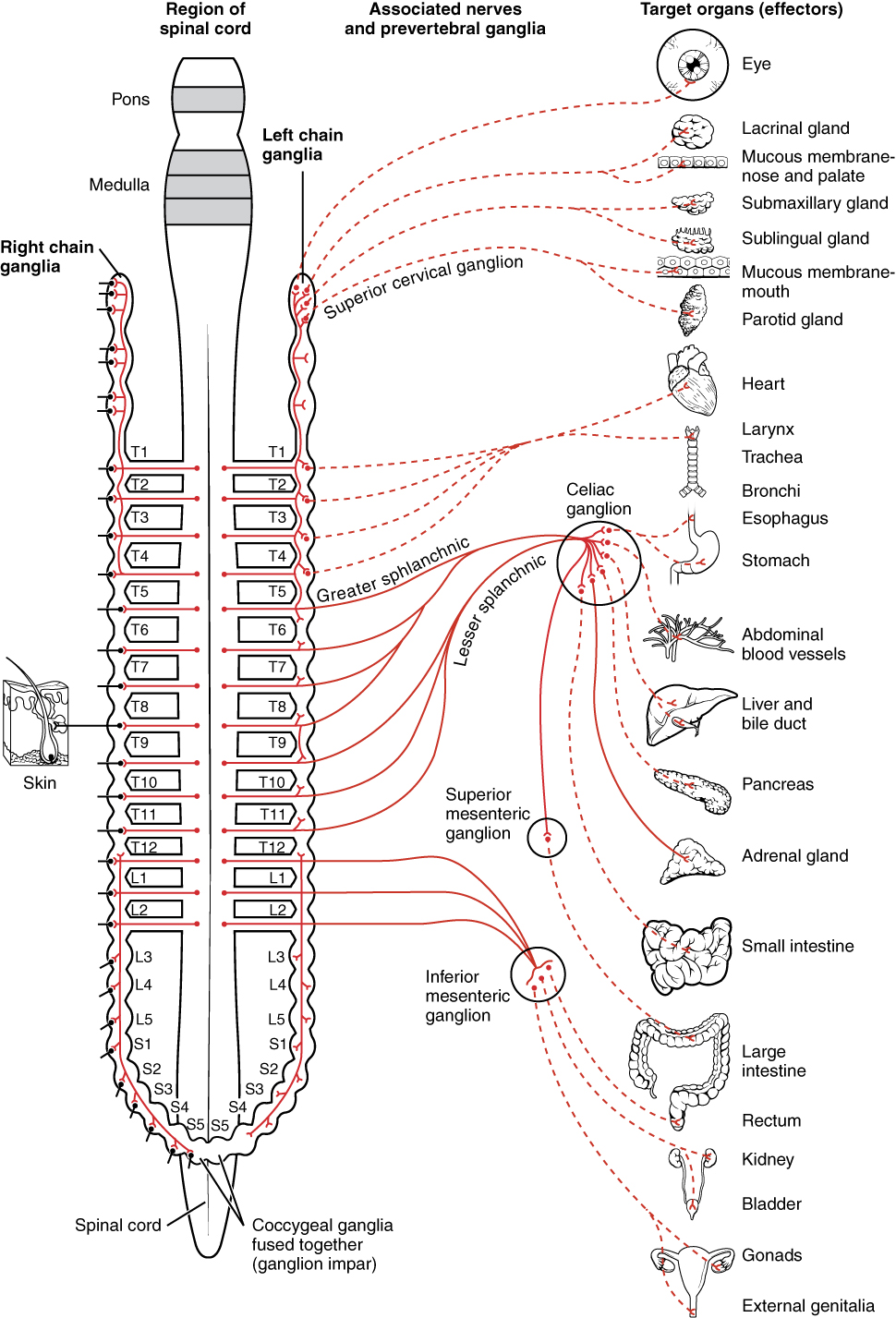
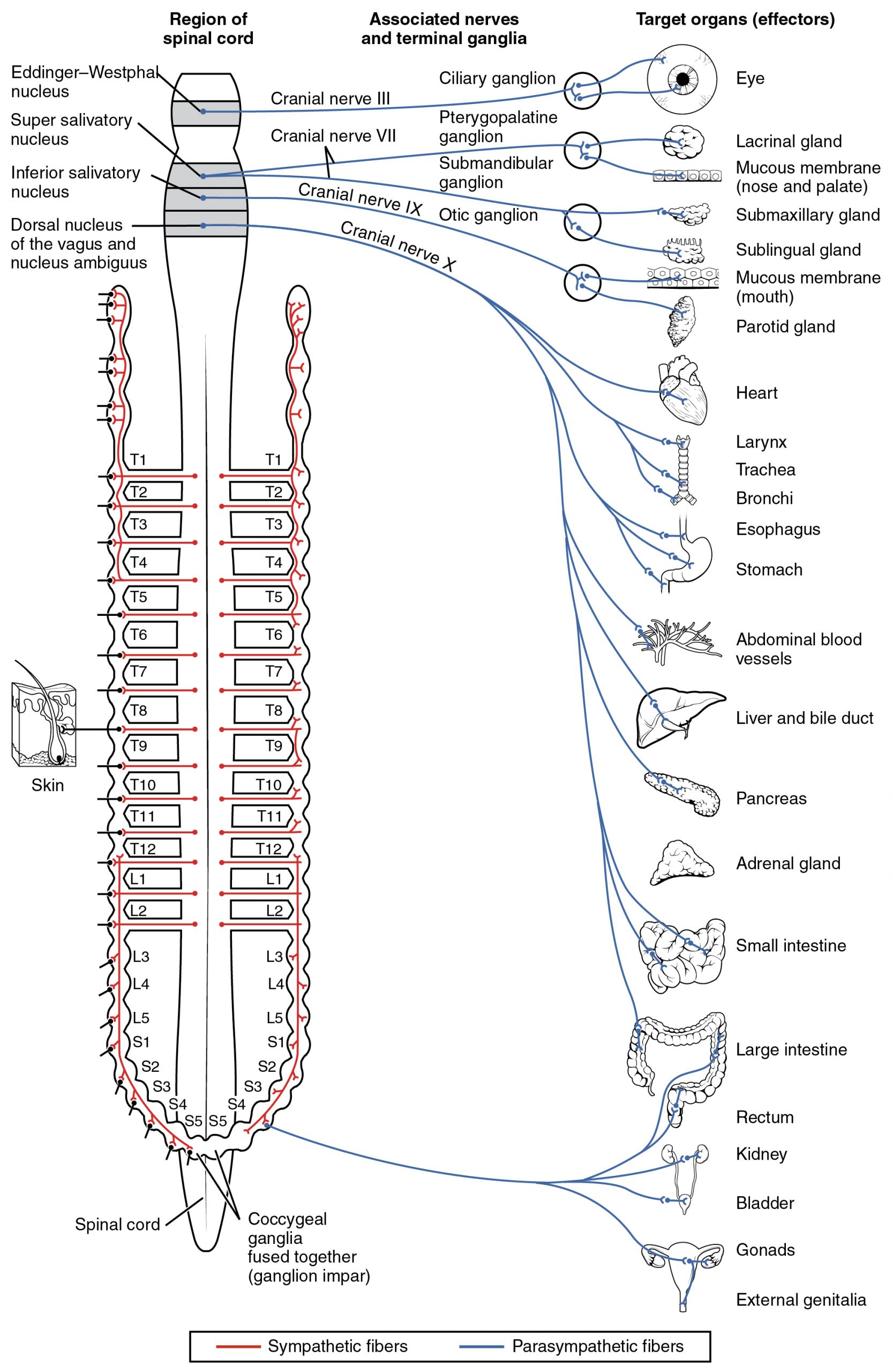
Sympathetic vs Parasympathetic
| Feature | Sympathetic ("Fight or Flight") | Parasympathetic ("Rest and Digest") |
|---|---|---|
| Origin | Thoracolumbar (T1-L2) spinal cord | Craniosacral (CN III, VII, IX, X; S2-S4) |
| Preganglionic NT | ACh (nicotinic receptors) | ACh (nicotinic receptors) |
| Postganglionic NT | Norepinephrine (adrenergic, except sweat glands → ACh) | ACh (muscarinic receptors) |
| Ganglia location | Close to spinal cord (paravertebral/prevertebral) | Close to or within target organ |
| Heart | Increased HR (beta-1), increased contractility | Decreased HR (M2), decreased AV conduction |
| Bronchi | Bronchodilation (beta-2) | Bronchoconstriction (M3) |
| Blood vessels | Vasoconstriction (alpha-1); vasodilation in skeletal muscle (beta-2) | Minimal direct effect (NO-mediated vasodilation in some beds) |
| GI tract | Decreased motility, sphincter contraction | Increased motility, sphincter relaxation |
| Pupils | Mydriasis (alpha-1, dilator muscle) | Miosis (M3, sphincter muscle) |
| Bladder | Relaxation of detrusor (beta-2), contraction of sphincter (alpha-1) | Contraction of detrusor (M3), relaxation of sphincter |
Adrenergic Receptor Subtypes
| Receptor | G-Protein | Primary Locations | Key Actions |
|---|---|---|---|
| Alpha-1 | Gq (IP3/DAG) | Vascular smooth muscle, iris dilator, bladder sphincter | Vasoconstriction, mydriasis, urinary retention |
| Alpha-2 | Gi (decrease cAMP) | Presynaptic nerve terminals, CNS | Inhibits NE release (negative feedback); clonidine agonist → lowers BP |
| Beta-1 | Gs (increase cAMP) | Heart, JG cells | Increased HR, contractility, conduction, renin release |
| Beta-2 | Gs (increase cAMP) | Bronchial smooth muscle, vascular smooth muscle, uterus | Bronchodilation, vasodilation, tocolysis, glycogenolysis |
| Beta-3 | Gs (increase cAMP) | Adipose tissue, bladder | Lipolysis, bladder relaxation (mirabegron) |
21 Sensory & Motor Systems
Spinal Cord Lesion Syndromes
| Syndrome | Lesion | Deficits | Classic Cause |
|---|---|---|---|
| Brown-Sequard | Hemisection of spinal cord | Ipsilateral: motor loss (corticospinal), vibration/proprioception loss (DCML). Contralateral: pain/temp loss (spinothalamic, 1-2 levels below) | Penetrating trauma, lateral mass lesion |
| Central cord | Central spinal cord (inner fibers) | Upper extremity > lower extremity weakness (cervical motor fibers are medial); bilateral loss of pain/temp at level of lesion (crossing spinothalamic fibers in anterior white commissure) | Syringomyelia, hyperextension injury in elderly with cervical spondylosis |
| Anterior cord | Anterior 2/3 of cord (anterior spinal artery) | Bilateral motor paralysis (corticospinal) and pain/temp loss (spinothalamic) below lesion; preserved vibration/proprioception (DCML, posterior columns intact) | Aortic dissection/surgery, anterior spinal artery occlusion |
| Posterior cord | Posterior columns | Bilateral loss of vibration, proprioception, fine touch; sensory ataxia | B12 deficiency (subacute combined degeneration), tabes dorsalis (syphilis) |
| Cauda equina | Nerve roots below L1-L2 | LMN signs: flaccid paralysis, areflexia, saddle anesthesia, bladder/bowel incontinence | Large disc herniation, tumor, epidural abscess |
Sensory Pathways
| Tract | Modality | First-Order Neuron | Decussation | Clinical Lesion |
|---|---|---|---|---|
| Dorsal columns (DCML) | Fine touch, vibration, proprioception | DRG → ipsilateral dorsal columns | Medulla (internal arcuate fibers) | Tabes dorsalis (syphilis), B12 deficiency, MS |
| Spinothalamic (anterolateral) | Pain, temperature, crude touch | DRG → dorsal horn | Anterior white commissure (1-2 levels above entry) | Syringomyelia (central cord cavitation → bilateral loss of pain/temp at level, preserved DCML) |
| Spinocerebellar | Unconscious proprioception | DRG → Clarke's nucleus / cuneocerebellum | Ipsilateral (mostly) | Friedreich ataxia |
Motor Pathways
The corticospinal (pyramidal) tract is the primary voluntary motor pathway. Upper motor neurons (UMN) originate in the primary motor cortex (precentral gyrus), descend through the internal capsule and cerebral peduncles, and 85-90% decussate at the pyramidal decussation in the medulla (forming the lateral corticospinal tract). They synapse on lower motor neurons (LMN) in the ventral horn of the spinal cord, which project to skeletal muscle via the ventral root.
| Finding | UMN Lesion | LMN Lesion |
|---|---|---|
| Muscle tone | Spasticity (clasp-knife) | Flaccidity |
| Reflexes | Hyperreflexia | Hyporeflexia/areflexia |
| Babinski sign | Present (upgoing plantar) | Absent (downgoing) |
| Atrophy | Mild, disuse | Significant, early |
| Fasciculations | Absent | Present |
| Distribution | Pyramidal pattern (extensors in UE, flexors in LE weaker) | Specific myotome/nerve distribution |
Cerebellum
The cerebellum coordinates movement, balance, and motor learning. Lesions cause ipsilateral deficits (cerebellar hemispheres control ipsilateral limbs because of double-crossing: corticopontocerebellar pathway crosses once, cerebellothalamocortical pathway crosses back). Signs: DANISH — Dysdiadochokinesia, Ataxia, Nystagmus, Intention tremor, Scanning speech, Hypotonia. Midline (vermis) lesions cause truncal ataxia and wide-based gait; lateral (hemisphere) lesions cause limb ataxia and dysmetria.
Basal Ganglia
The basal ganglia modulate movement through two parallel pathways: the direct pathway (facilitates movement: cortex → striatum [D1 receptors, excitatory] → inhibits GPi/SNr → disinhibits thalamus → increases cortical motor output) and the indirect pathway (inhibits movement: cortex → striatum [D2 receptors, inhibitory] → less inhibition of GPe → more inhibition of STN → less excitation of GPi/SNr → more inhibition of thalamus → decreased cortical output). Dopamine from the substantia nigra pars compacta (SNc) activates the direct pathway (D1) and inhibits the indirect pathway (D2) — net effect: facilitates movement. In Parkinson disease, loss of dopaminergic neurons in the SNc leads to reduced direct pathway activity and increased indirect pathway activity → bradykinesia, rigidity, resting tremor. In Huntington disease, degeneration of striatal neurons (indirect pathway first) → excessive movement (chorea).
Special Senses — Vision
Phototransduction occurs in rods (dim light, peripheral vision, rhodopsin pigment) and cones (color vision, central vision — three types: S/blue, M/green, L/red). In the dark, retinal photoreceptors are depolarized (cGMP opens Na+ channels — "dark current") and continuously release glutamate. Light activates rhodopsin → transducin (Gt) → phosphodiesterase (PDE) → decreases cGMP → Na+ channels close → hyperpolarization → decreased glutamate release. This is a unique case where the stimulus (light) causes hyperpolarization, not depolarization. Sildenafil (Viagra) inhibits PDE5 (vascular smooth muscle) but can cross-inhibit retinal PDE6, causing blue-tinged vision (cyanopsia).
Special Senses — Hearing
Sound waves vibrate the tympanic membrane → ossicular chain (malleus, incus, stapes — amplifies pressure 20-fold by concentrating force from the large TM to the small oval window) → perilymph waves in the cochlea → basilar membrane vibration → deflection of stereocilia on inner hair cells → mechanotransduction channels open (K+ influx from K+-rich endolymph) → depolarization → glutamate release → CN VIII firing. The basilar membrane is tonotopically organized: base (narrow, stiff) encodes high frequencies; apex (wide, flexible) encodes low frequencies. Conductive hearing loss: middle ear pathology (otitis media, otosclerosis); Weber lateralizes to affected ear, Rinne shows bone > air. Sensorineural hearing loss: inner ear or CN VIII pathology (presbycusis, acoustic neuroma, ototoxic drugs); Weber lateralizes to unaffected ear, Rinne shows air > bone bilaterally.
22 Endocrine Physiology & Hormone Signaling
Hormone Classification by Mechanism
| Class | Examples | Receptor Location | Signal Transduction | Speed of Action |
|---|---|---|---|---|
| Peptide/Protein | Insulin, glucagon, GH, PTH, ADH | Cell surface (GPCR or RTK) | cAMP, IP3/DAG, JAK-STAT, RTK | Fast (minutes) |
| Steroid | Cortisol, aldosterone, testosterone, estradiol | Intracellular (nuclear) | Gene transcription via hormone-response elements | Slow (hours-days) |
| Thyroid | T3, T4 | Intracellular (nuclear) | Gene transcription (acts like steroid despite amine origin) | Slow (hours-days) |
| Amine (catecholamine) | Epinephrine, norepinephrine, dopamine | Cell surface (GPCR) | cAMP, IP3/DAG | Fast (seconds) |
Second Messenger Systems
cAMP pathway (Gs activates adenylyl cyclase → cAMP → PKA): hormones using this pathway include ACTH, TSH, LH, FSH, ADH (V2), PTH, glucagon, beta-adrenergic agonists. Gi inhibits adenylyl cyclase (alpha-2, M2). IP3/DAG pathway (Gq activates PLC → IP3 releases Ca2+ from ER; DAG activates PKC): hormones include Ang II, ADH (V1), oxytocin, GnRH, TRH, alpha-1 adrenergic. Receptor tyrosine kinase (RTK): insulin, IGF-1, EGF, PDGF — receptor autophosphorylation activates MAP kinase and PI3K/Akt pathways. JAK-STAT: GH, prolactin, erythropoietin, cytokines.
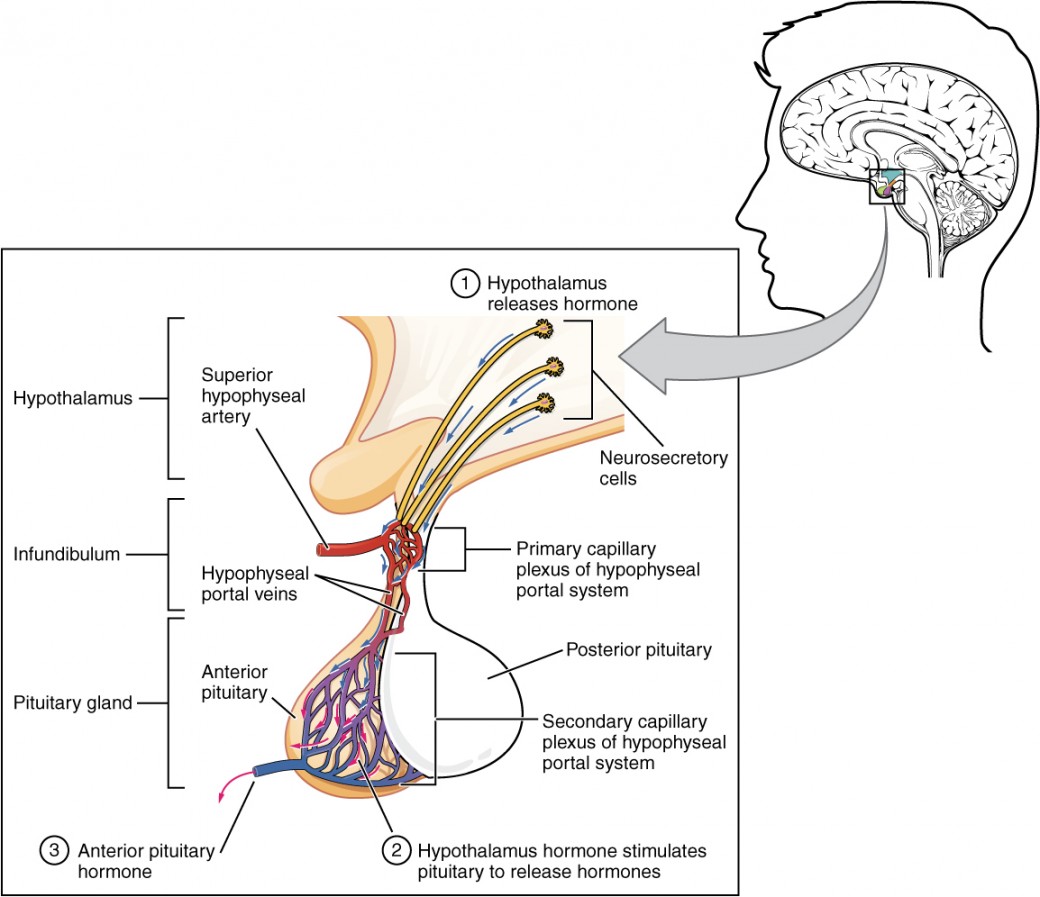
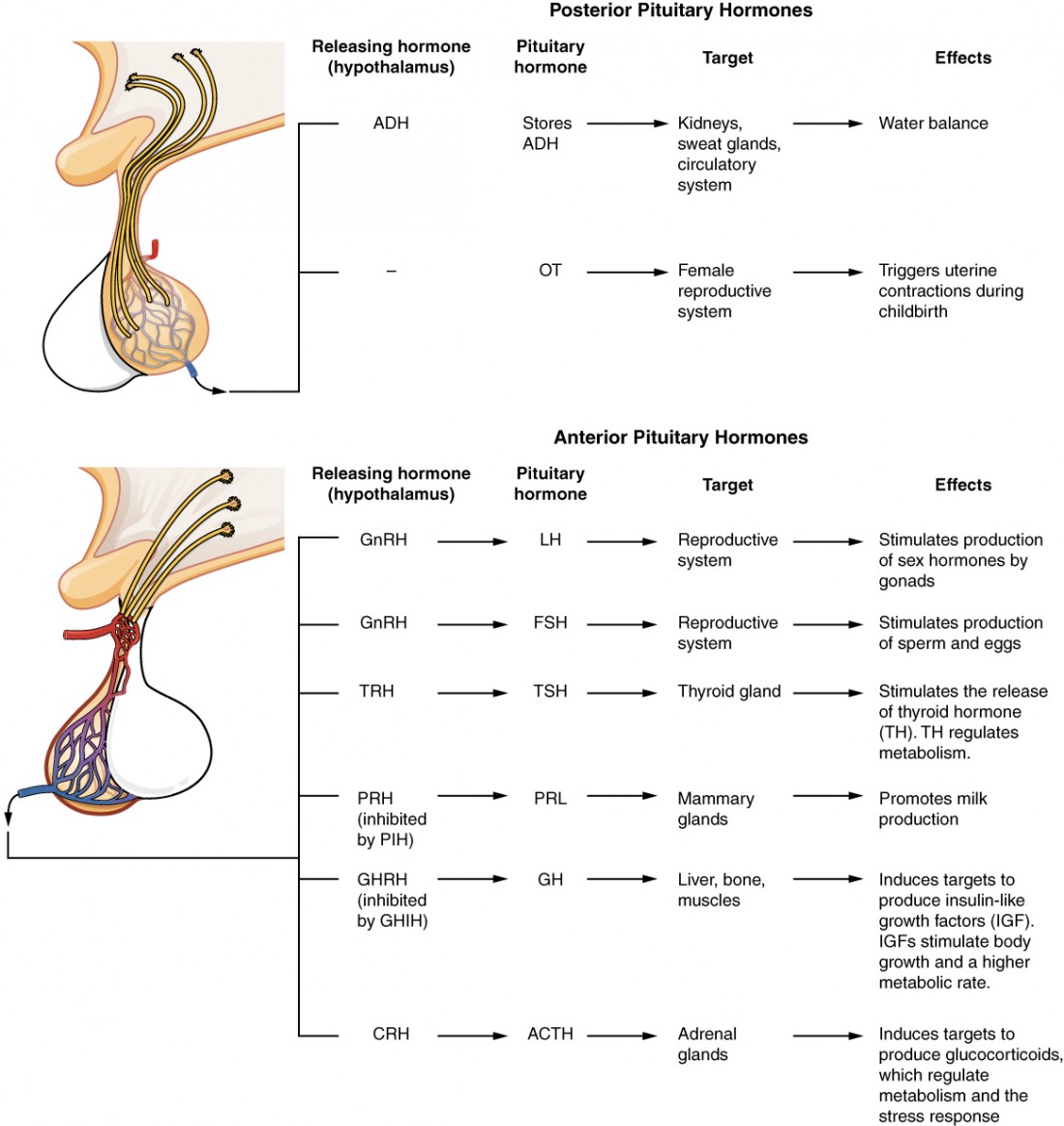
The Hypothalamic-Pituitary Axes
| Hypothalamic Hormone | Pituitary Hormone | Target & End Hormone | Feedback |
|---|---|---|---|
| CRH | ACTH (anterior) | Adrenal cortex → Cortisol | Cortisol inhibits CRH + ACTH |
| TRH | TSH (anterior) | Thyroid → T3/T4 | T3/T4 inhibit TRH + TSH |
| GnRH (pulsatile) | LH, FSH (anterior) | Gonads → Testosterone/Estradiol | Sex steroids inhibit GnRH + LH/FSH |
| GHRH / Somatostatin | GH (anterior) | Liver → IGF-1 | IGF-1 and GH inhibit GHRH, stimulate somatostatin |
| Dopamine (inhibitory) | Prolactin (anterior) | Breast → lactation | Prolactin stimulates dopamine (short-loop feedback) |
Insulin & Glucagon — Glucose Homeostasis
Insulin (from pancreatic beta cells) is the primary anabolic hormone. It is released in response to elevated blood glucose (glucose enters beta cells via GLUT2 → glycolysis → increased ATP → closes KATP channels → depolarization → Ca2+ influx → insulin vesicle exocytosis). Insulin promotes glucose uptake (via GLUT4 translocation in muscle and adipose), glycogenesis, lipogenesis, and protein synthesis. It suppresses gluconeogenesis, glycogenolysis, lipolysis, and proteolysis. Insulin signals through a receptor tyrosine kinase (RTK) → IRS-1 → PI3K/Akt pathway → GLUT4 insertion and metabolic effects.
Glucagon (from pancreatic alpha cells) is the counterregulatory hormone, released during hypoglycemia. It acts via Gs-coupled receptors → cAMP → PKA in hepatocytes to promote glycogenolysis and gluconeogenesis, raising blood glucose. Other counterregulatory hormones include epinephrine, cortisol, and GH — all are "diabetogenic" (raise glucose). In type 1 diabetes, loss of insulin AND impaired glucagon response to hypoglycemia (from loss of paracrine insulin signaling to alpha cells) creates "hypoglycemia unawareness" — a dangerous complication.
| State | Insulin | Glucagon | Metabolic Effect |
|---|---|---|---|
| Fed (postprandial) | High | Low | Glucose uptake, glycogen synthesis, lipogenesis, protein synthesis |
| Fasting (4-16 hours) | Low | High | Glycogenolysis (primary), early gluconeogenesis |
| Starvation (days) | Very low | High | Gluconeogenesis (from amino acids, glycerol, lactate), lipolysis → ketogenesis |
| Exercise | Low | High + epinephrine | Glycogenolysis, lipolysis; GLUT4 translocation via AMPK (insulin-independent) |
23 Thyroid, Adrenal & Calcium Homeostasis
Thyroid Physiology
The thyroid gland synthesizes T4 (thyroxine, ~90% of output, relatively inactive) and T3 (triiodothyronine, ~10% of output, 3-5x more active). Peripheral conversion of T4 to T3 by 5'-deiodinase (type 1 and 2) produces most circulating T3. Type 3 deiodinase converts T4 to reverse T3 (rT3), which is inactive — increased in critical illness ("sick euthyroid" or "euthyroid sick syndrome"). Thyroid hormones are lipophilic and travel in blood mostly bound to TBG (thyroxine-binding globulin); only the free fraction is biologically active.
Thyroid hormones increase basal metabolic rate, oxygen consumption, heat production, heart rate and contractility (upregulate beta-1 receptors), and are essential for CNS development in neonates (cretinism if deficient). Wolff-Chaikoff effect: excess iodine transiently inhibits thyroid hormone synthesis (used acutely in thyroid storm). Jod-Basedow effect: excess iodine triggers hyperthyroidism in patients with underlying thyroid autonomy (toxic nodular goiter).
Thyroid Function Tests — Interpretation
| TSH | Free T4 | Diagnosis | Common Causes |
|---|---|---|---|
| Low | High | Primary hyperthyroidism | Graves disease, toxic nodular goiter, thyroiditis (early) |
| High | Low | Primary hypothyroidism | Hashimoto thyroiditis, post-ablation, iodine deficiency |
| Low | Low | Secondary (central) hypothyroidism | Pituitary adenoma, Sheehan syndrome, pituitary surgery |
| High | Normal | Subclinical hypothyroidism | Early Hashimoto, incomplete replacement |
| Low | Normal | Subclinical hyperthyroidism | Toxic nodule, excessive levothyroxine dose |
| Normal/High | High | TSH-secreting pituitary adenoma or thyroid hormone resistance | Rare; inappropriate TSH secretion |
Adrenal Physiology
The adrenal cortex produces steroids in three zones (mnemonic: "GFR — Salt, Sugar, Sex"): zona Glomerulosa (aldosterone — mineralocorticoid, regulated by Ang II and K+), zona Fasciculata (cortisol — glucocorticoid, regulated by ACTH), zona Reticularis (DHEA/androgens, regulated by ACTH). The adrenal medulla produces catecholamines (epinephrine 80%, norepinephrine 20%).
Cortisol actions: increased gluconeogenesis, lipolysis, proteolysis (catabolic); anti-inflammatory and immunosuppressive; maintains vascular tone (permissive effect on catecholamines); inhibits bone formation. Cortisol follows a diurnal rhythm (peak 6-8 AM, nadir at midnight). Chronic cortisol excess (Cushing syndrome) causes central obesity, striae, hyperglycemia, osteoporosis, proximal myopathy, and immunosuppression.
Adrenal Insufficiency & Stress Response
The adrenal cortex is essential for the stress response. Primary adrenal insufficiency (Addison disease): destruction of the adrenal cortex (autoimmune most common in developed countries, TB worldwide) → deficiency of cortisol AND aldosterone. Presents with hypotension, hyperkalemia, metabolic acidosis, hyperpigmentation (increased ACTH from loss of negative feedback → ACTH is cleaved from POMC, which also yields MSH). Secondary adrenal insufficiency: decreased ACTH (most commonly from chronic exogenous glucocorticoid use → hypothalamic-pituitary suppression → adrenal atrophy). No hyperpigmentation (ACTH is low). Aldosterone is usually preserved (regulated mainly by RAAS and K+, not ACTH). Adrenal crisis is a life-threatening emergency precipitated by physiologic stress (surgery, infection) in a patient with unrecognized adrenal insufficiency or abrupt steroid withdrawal. Treatment: IV hydrocortisone (stress dose = 100 mg bolus then 50 mg q8h) + aggressive fluid resuscitation.
Cholesterol → Pregnenolone (rate-limiting step, via desmolase/CYP11A1) → branches into:
Mineralocorticoid pathway: Pregnenolone → Progesterone → 11-Deoxycorticosterone → Corticosterone → Aldosterone (zona glomerulosa, requires aldosterone synthase).
Glucocorticoid pathway: Pregnenolone → 17-OH Pregnenolone → 17-OH Progesterone → 11-Deoxycortisol → Cortisol (zona fasciculata, requires 11-beta-hydroxylase and 17-alpha-hydroxylase).
Androgen pathway: 17-OH Pregnenolone → DHEA → Androstenedione → Testosterone (zona reticularis).
Enzyme deficiencies cause congenital adrenal hyperplasia (CAH): 21-hydroxylase deficiency (most common, 90%) → cannot make cortisol or aldosterone → ACTH-driven androgen excess → virilization in females, salt-wasting in severe form.
Calcium Homeostasis
| Hormone | Source | Stimulus | Effect on Ca2+ | Mechanism |
|---|---|---|---|---|
| PTH | Parathyroid glands (chief cells) | Low ionized Ca2+ (via CaSR) | Increases serum Ca2+ | Increases bone resorption (osteoclasts), increases renal Ca2+ reabsorption (DCT), increases 1-alpha-hydroxylase (more active vitamin D), decreases PO43- reabsorption (phosphaturic) |
| Vitamin D (1,25-dihydroxy) | Kidney (1-alpha-hydroxylase converts 25-OH-D) | PTH, low PO43- | Increases serum Ca2+ and PO43- | Increases intestinal Ca2+ and PO43- absorption; enhances bone mineralization |
| Calcitonin | Thyroid parafollicular C cells | High serum Ca2+ | Decreases serum Ca2+ (minor role) | Inhibits osteoclast activity; tones down bone resorption |
PTH: "Phosphate Trashing Hormone" — raises Ca2+, lowers PO43- (phosphaturic).
Vitamin D: Raises BOTH Ca2+ and PO43- (increases intestinal absorption of both).
Calcitonin: "Calci-tone-it-down" — lowers Ca2+ (minor physiologic significance; tumor marker for medullary thyroid cancer).
24 Muscle Physiology & Excitation-Contraction Coupling
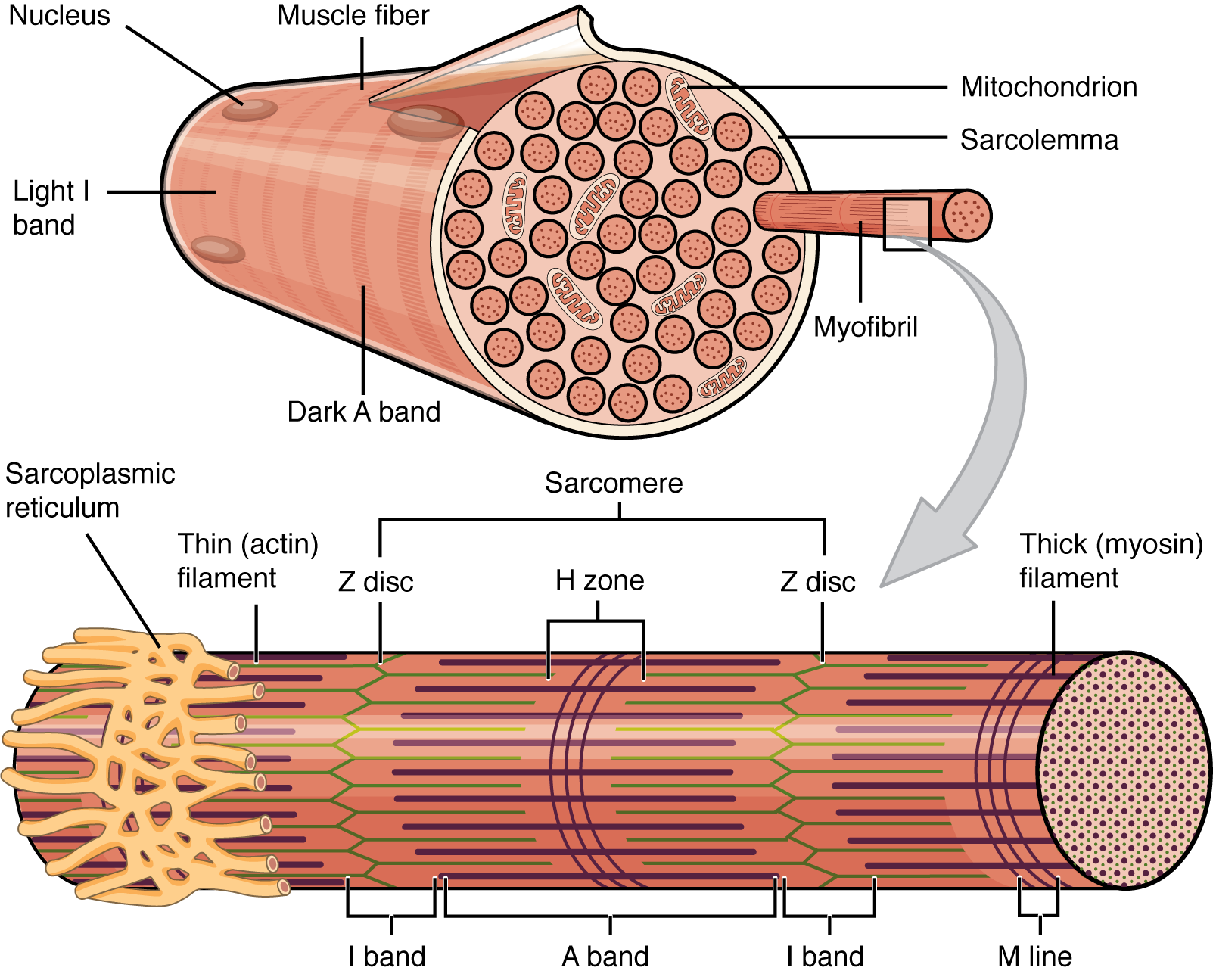
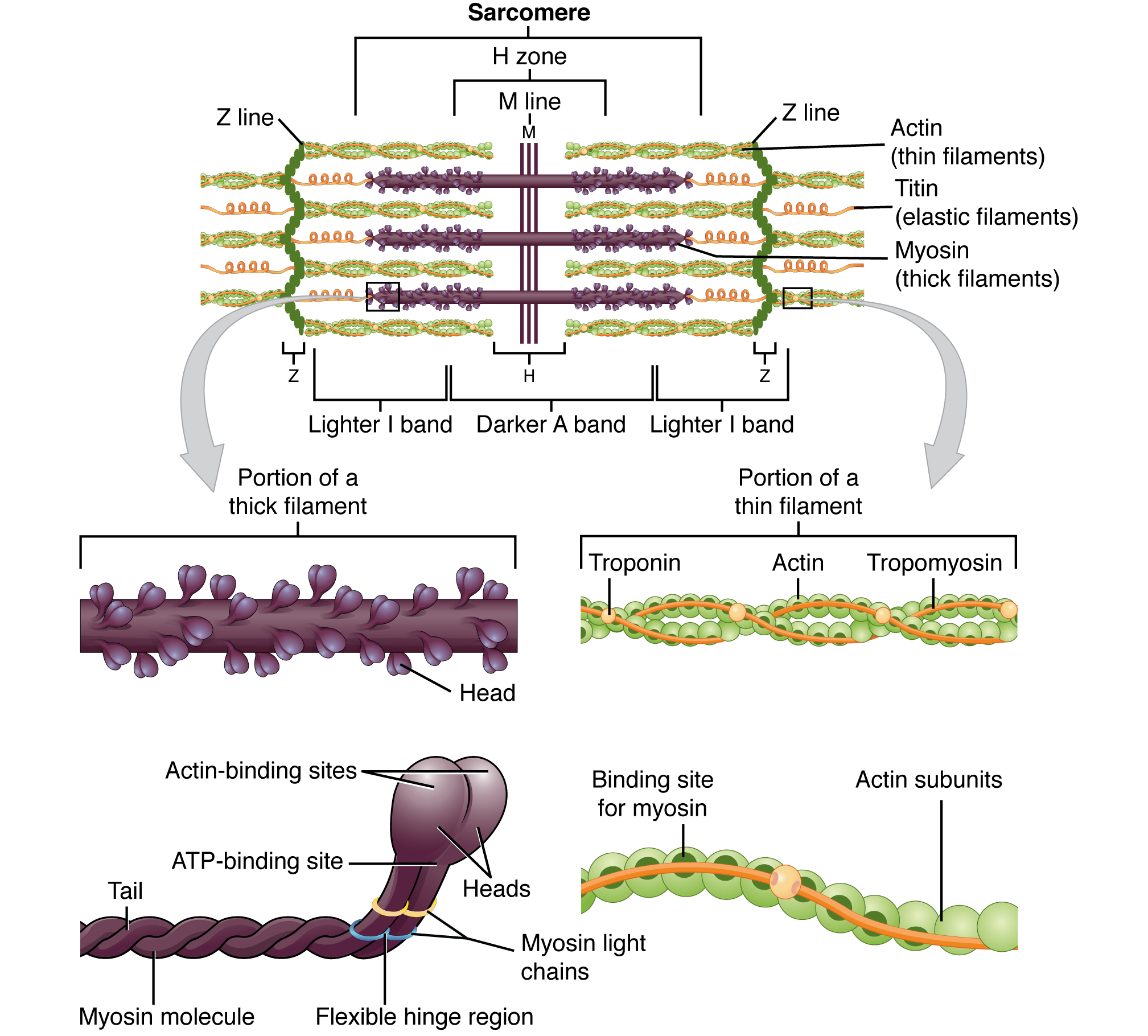
Skeletal Muscle Contraction
The sliding filament theory: muscle contraction occurs when actin thin filaments slide past myosin thick filaments, shortening the sarcomere. The sequence: (1) AP at the NMJ → ACh release → nicotinic receptor activation → end-plate potential → muscle fiber AP, (2) AP propagates along the sarcolemma and down T-tubules, (3) voltage change in T-tubules activates the dihydropyridine receptor (DHPR), which is mechanically coupled to the ryanodine receptor (RyR1) on the sarcoplasmic reticulum (SR), (4) Ca2+ release from SR binds troponin C → conformational change moves tropomyosin off the actin binding sites → myosin cross-bridge cycling begins (requires ATP), (5) relaxation occurs when Ca2+ is pumped back into the SR by SERCA (SR Ca2+-ATPase).
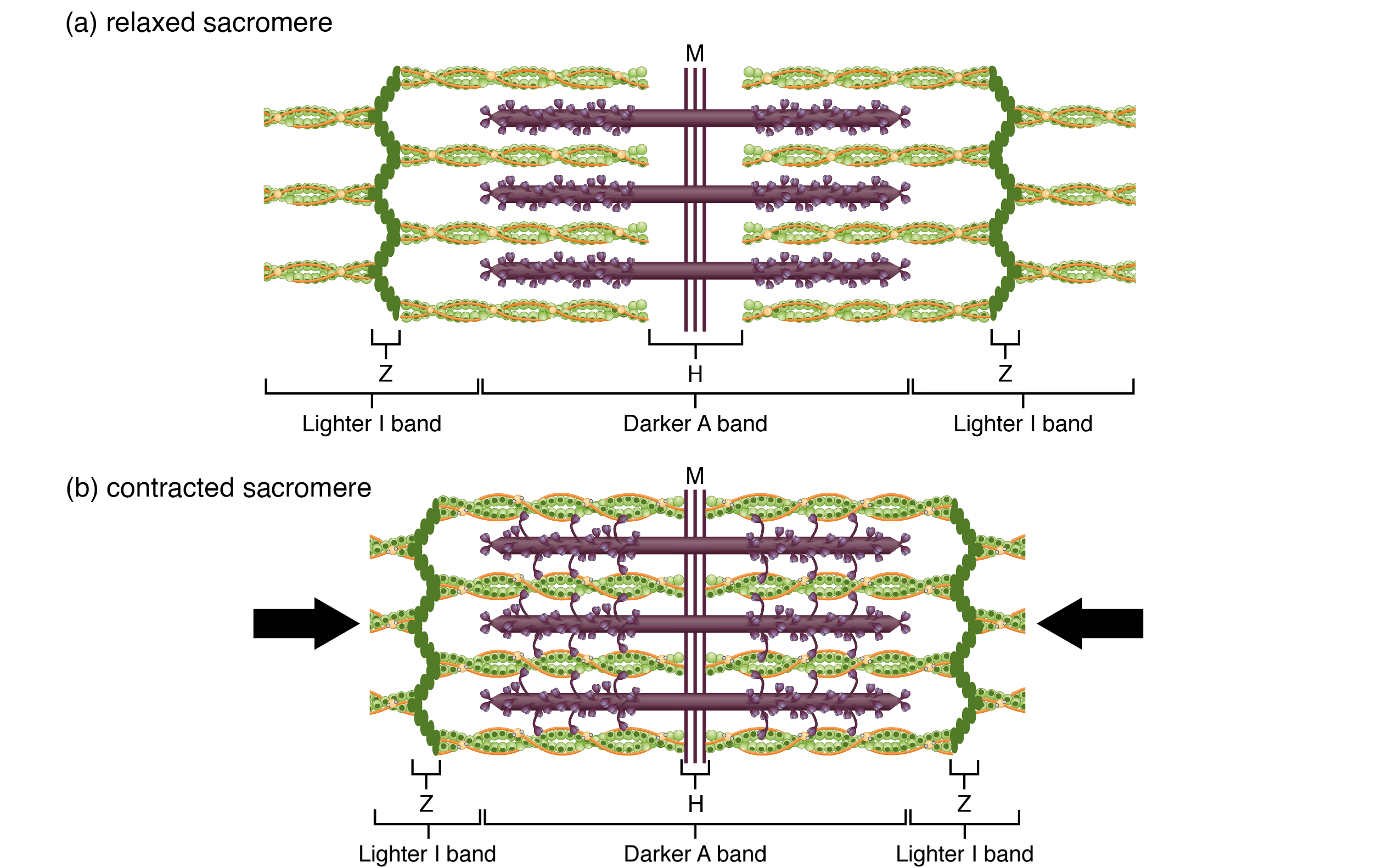
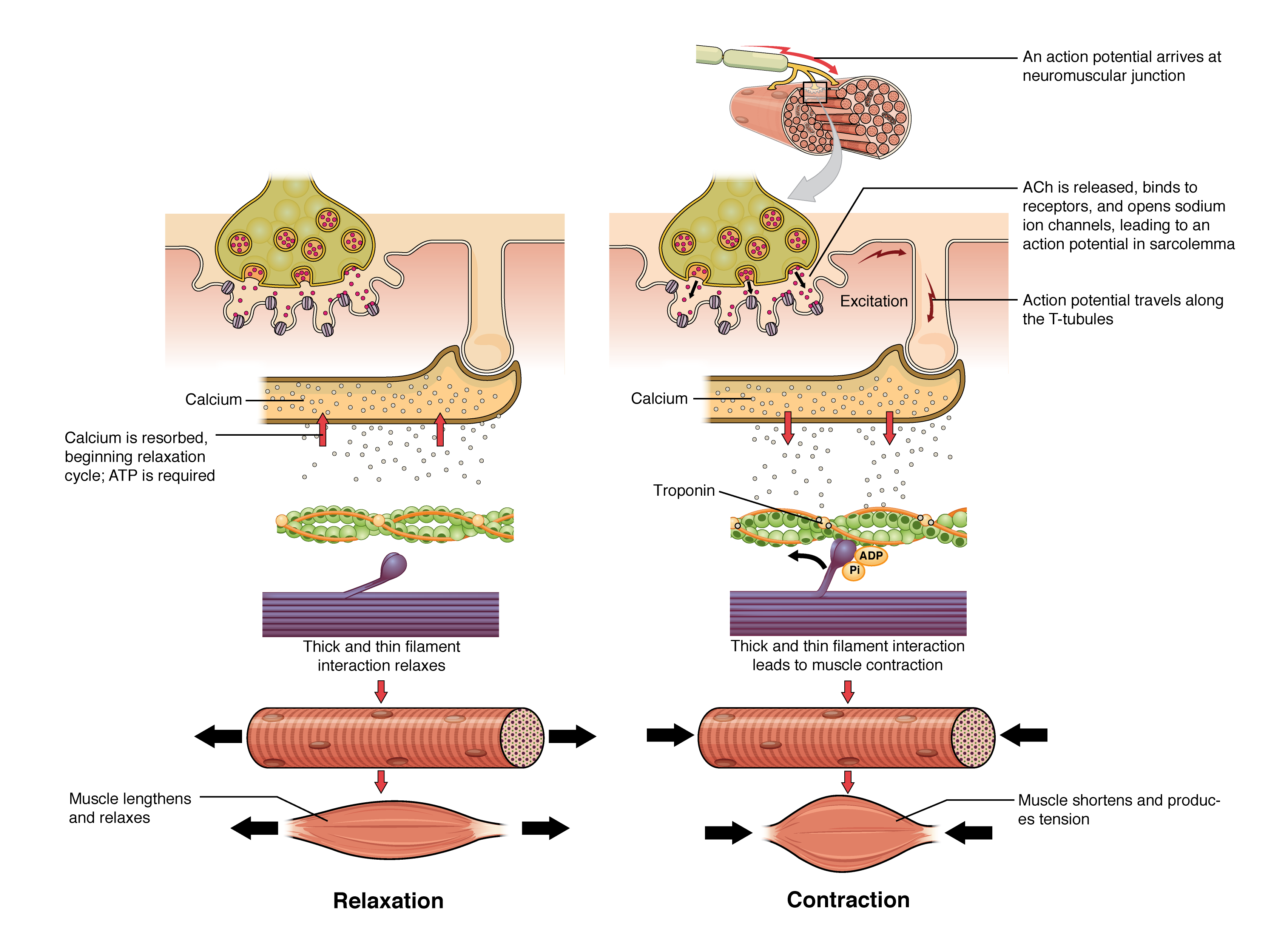
Muscle Fiber Types
| Feature | Type I (Slow Oxidative) | Type IIa (Fast Oxidative) | Type IIb (Fast Glycolytic) |
|---|---|---|---|
| Color | Red (high myoglobin) | Red-pink | White (low myoglobin) |
| Metabolism | Aerobic (oxidative phosphorylation) | Aerobic + anaerobic | Anaerobic (glycolysis) |
| Fatigue resistance | High | Moderate | Low |
| Mitochondria | Many | Many | Few |
| Function | Posture, endurance | Walking, sustained activity | Sprinting, power movements |
Cardiac vs Skeletal vs Smooth Muscle
| Feature | Skeletal | Cardiac | Smooth |
|---|---|---|---|
| Striated | Yes | Yes | No |
| Voluntary | Yes | No | No |
| Nuclei | Multinucleated, peripheral | 1-2, central | 1, central |
| EC coupling | DHPR-RyR1 mechanical coupling | Ca2+-induced Ca2+ release (CICR) via RyR2 | IP3-mediated Ca2+ release + extracellular Ca2+ influx |
| Regulatory protein | Troponin-tropomyosin | Troponin-tropomyosin | Calmodulin → MLCK |
| Gap junctions | No | Yes (intercalated discs) | Yes (single-unit type) |
| Tetanus possible | Yes | No (long refractory period) | Yes |
Length-Tension & Force-Velocity Relationships
The length-tension relationship describes how muscle force varies with sarcomere length. Maximum active tension is generated at optimal sarcomere length (~2.2 micrometers) where actin-myosin overlap is maximal. Stretching beyond optimal length reduces overlap and force (descending limb). Excessive shortening causes thin filament overlap and reduced force (ascending limb). In the heart, the ascending limb of this curve is the physiologic basis of the Frank-Starling mechanism — increased ventricular filling (preload) stretches sarcomeres toward optimal length, increasing contractile force.
The force-velocity relationship shows that as afterload (force) increases, shortening velocity decreases. At maximum isometric force (very high afterload), velocity is zero (isovolumetric contraction). At zero load, velocity is maximum (unloaded shortening — Vmax). Vmax reflects intrinsic contractility independent of loading conditions. Positive inotropes (catecholamines, digoxin) increase Vmax; heart failure decreases Vmax.
Smooth Muscle Contraction
Unlike skeletal and cardiac muscle (which use troponin-tropomyosin), smooth muscle contraction is regulated by calmodulin-MLCK. When Ca2+ rises (from IP3-mediated release from SR or extracellular influx through L-type Ca2+ channels), it binds calmodulin → activates myosin light chain kinase (MLCK) → phosphorylates myosin light chain → allows actin-myosin cross-bridge cycling → contraction. Relaxation occurs when Ca2+ falls and myosin light chain phosphatase (MLCP) dephosphorylates myosin. NO (nitric oxide) causes smooth muscle relaxation by activating guanylyl cyclase → increased cGMP → activates PKG → decreases intracellular Ca2+ and activates MLCP. This is the mechanism of nitroglycerin-mediated vasodilation. PDE5 inhibitors (sildenafil) prevent cGMP degradation, prolonging the vasodilatory effect — used for erectile dysfunction and pulmonary hypertension.
25 Reproductive Physiology
Growth Hormone & Prolactin
Growth hormone (GH) is released from anterior pituitary somatotrophs in a pulsatile fashion (peak during deep sleep). It is stimulated by GHRH and ghrelin, inhibited by somatostatin and IGF-1. GH acts directly (lipolysis, insulin resistance, gluconeogenesis) and indirectly via IGF-1 (produced by the liver — promotes linear growth at epiphyseal plates, protein synthesis, and cell proliferation). GH excess before epiphyseal closure → gigantism; after closure → acromegaly (enlarged hands, feet, jaw, visceral organs, carpal tunnel, diabetes). GH deficiency in children → short stature; in adults → decreased lean mass, increased fat, fatigue.
Prolactin is the only anterior pituitary hormone under predominantly inhibitory control (by dopamine from the hypothalamus via the tuberoinfundibular pathway). Prolactin stimulates breast milk production. It is increased by: pregnancy, breastfeeding (suckling reflex), prolactinomas, dopamine antagonists (antipsychotics, metoclopramide), hypothyroidism (TRH stimulates prolactin release). High prolactin inhibits GnRH → hypogonadism (amenorrhea, infertility, decreased libido). Treatment of prolactinoma: dopamine agonists (cabergoline, bromocriptine) — first-line even for macroprolactinomas; surgery is rarely needed.
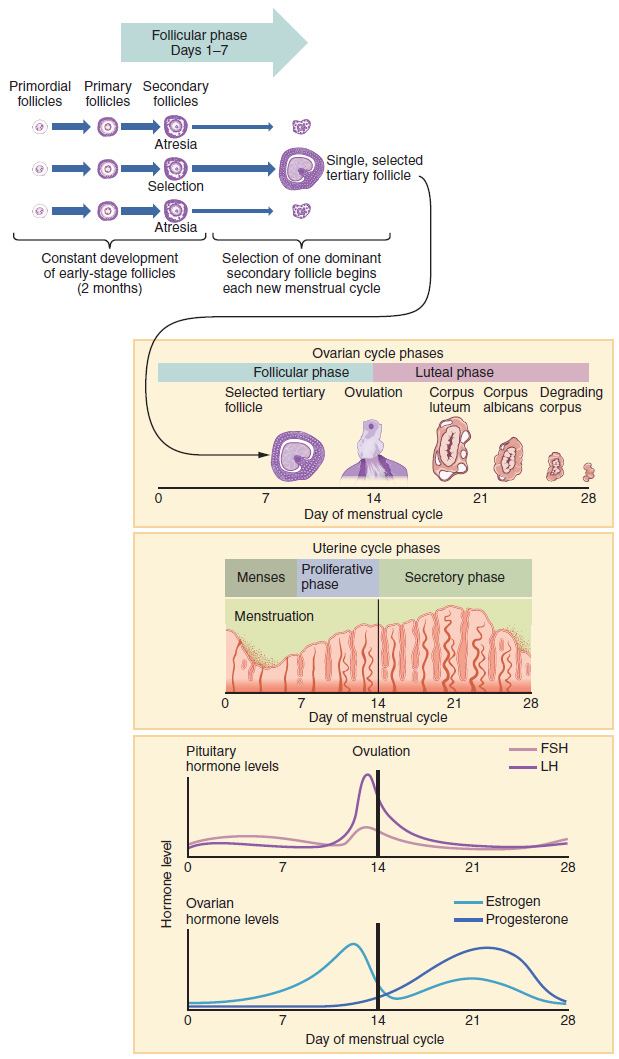
Fetal Physiology & Transitional Circulation
In utero, gas exchange occurs at the placenta (not the lungs). Fetal circulation features three shunts: (1) ductus venosus (bypasses the liver, shunting oxygenated blood from the umbilical vein to the IVC), (2) foramen ovale (right-to-left shunt from RA to LA, bypassing the lungs), (3) ductus arteriosus (right-to-left shunt from pulmonary artery to aorta, bypassing the lungs). These shunts exist because fetal pulmonary vascular resistance is very high (fluid-filled lungs, hypoxic vasoconstriction). At birth: first breath expands lungs → decreased pulmonary resistance → increased pulmonary blood flow → increased LA pressure → functional closure of foramen ovale. Rising PaO2 + falling prostaglandins → closure of ductus arteriosus (indomethacin accelerates closure; PGE1 keeps it open in ductal-dependent congenital heart disease).
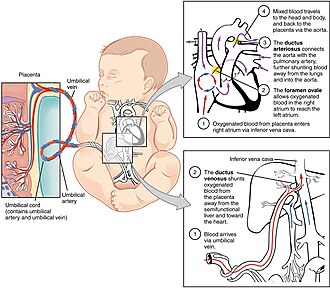
Male Reproductive Physiology
The hypothalamic-pituitary-gonadal (HPG) axis drives spermatogenesis and testosterone production. GnRH (pulsatile) stimulates LH and FSH from anterior pituitary gonadotrophs. LH acts on Leydig cells to produce testosterone. FSH acts on Sertoli cells to support spermatogenesis (Sertoli cells also produce inhibin B, which provides negative feedback on FSH, and androgen-binding protein, which maintains high local testosterone). Testosterone is converted to DHT (by 5-alpha-reductase — more potent androgen responsible for external genitalia development, prostate growth, male-pattern hair) and to estradiol (by aromatase — bone epiphyseal closure, feedback on HPG axis). Spermatogenesis takes ~74 days and requires the lower scrotal temperature (~2-3 degrees C below core body temperature).
Female Reproductive Physiology — The Menstrual Cycle
The average menstrual cycle is 28 days. Follicular phase (days 1-14): FSH stimulates follicular growth; granulosa cells produce estradiol; one dominant follicle emerges. Rising estradiol initially has negative feedback on GnRH/LH/FSH. When estradiol reaches a sustained high threshold (greater than 200 pg/mL for more than 36 hours), it switches to positive feedback, triggering the LH surge (~day 12). Ovulation occurs ~36 hours after the LH surge. Luteal phase (days 14-28): the corpus luteum produces progesterone (and estradiol), which stabilizes the endometrium for implantation, raises basal body temperature, and provides negative feedback on LH/FSH. If no implantation occurs, the corpus luteum degenerates → progesterone drops → endometrial shedding (menses). If implantation occurs, hCG from the trophoblast maintains the corpus luteum until the placenta takes over steroid production (~8-10 weeks).
Combined OCPs (estrogen + progestin) suppress the LH surge (preventing ovulation), thicken cervical mucus, and thin the endometrium. Progestin-only methods (mini-pill, implant, hormonal IUD) primarily work via cervical mucus thickening and endometrial thinning; they variably suppress ovulation. Emergency contraception (ulipristal, high-dose levonorgestrel) delays or inhibits ovulation; copper IUD prevents fertilization and implantation.
Pregnancy Hormones
| Hormone | Source | Peak Timing | Function |
|---|---|---|---|
| hCG | Syncytiotrophoblast | 8-10 weeks (then declines) | Maintains corpus luteum → progesterone production; basis of pregnancy testing |
| Progesterone | Corpus luteum (early), placenta (after 8-10 wk) | Rises throughout pregnancy | Maintains endometrium, inhibits uterine contractions, immunosuppression |
| Estrogen (estriol E3) | Placenta (from fetal DHEA-S precursors) | Rises throughout pregnancy | Uterine growth, breast development, increased hepatic protein synthesis |
| hPL (human placental lactogen) | Syncytiotrophoblast | Third trimester | Insulin resistance (ensures glucose delivery to fetus), lipolysis, breast development |
| Relaxin | Corpus luteum, placenta | First trimester peak | Softens cervix, relaxes pelvic ligaments, inhibits uterine contractions |
26 Clinical Correlates & Integrative Physiology
Fluid Compartments & Body Water Distribution
Total body water (TBW) = ~60% of body weight in a 70 kg male (~42 L). Distribution: intracellular fluid (ICF) = 2/3 of TBW (~28 L); extracellular fluid (ECF) = 1/3 of TBW (~14 L). ECF is further divided into interstitial fluid (3/4 of ECF, ~10.5 L) and plasma (1/4 of ECF, ~3.5 L). Measurement: TBW = tritiated water or deuterium oxide distribution; ECF = inulin or mannitol distribution; plasma volume = radiolabeled albumin or Evans blue dye; ICF = TBW - ECF (calculated).
| Fluid Added | Osmolarity Change | ECF Volume | ICF Volume | Clinical Example |
|---|---|---|---|---|
| Isotonic NaCl (0.9%) | No change | Increases | No change | Volume resuscitation |
| Hypertonic NaCl (3%) | Increases | Increases (more) | Decreases (water shift out) | Severe hyponatremia with seizures |
| Hypotonic fluid (D5W) | Decreases | Increases (slightly) | Increases (more) | Free water excess; distributes across TBW |
| Pure water loss (insensible) | Increases | Decreases | Decreases (more) | Diabetes insipidus, fever |
| Isotonic fluid loss | No change | Decreases | No change | Hemorrhage, diarrhea |
Exercise Physiology
During exercise, the integrated cardiovascular, respiratory, and metabolic responses include: increased sympathetic tone → increased HR, contractility, and venous return → increased CO (up to 5-fold). SVR decreases (beta-2 vasodilation in skeletal muscle outweighs alpha-1 vasoconstriction in splanchnic/renal beds). SBP increases; DBP stays constant or slightly decreases. Minute ventilation increases (up to 20-fold) through both rate and depth. The oxygen-hemoglobin dissociation curve right-shifts (increased temperature, CO2, 2,3-DPG, and lactic acid) to enhance O2 delivery. At the anaerobic threshold, lactate production exceeds clearance, causing metabolic acidosis and increased ventilatory drive.
Shock Classification — Integrative Physiology
| Type | Mechanism | Examples | Key Physiology |
|---|---|---|---|
| Hypovolemic | Decreased preload (volume loss) | Hemorrhage, dehydration, burns | Decreased venous return → decreased EDV → decreased CO (Starling) |
| Cardiogenic | Pump failure | MI, myocarditis, arrhythmia, valvular disease | Decreased contractility → increased ESV → decreased SV despite adequate preload |
| Distributive | Decreased SVR (vasodilation) | Sepsis, anaphylaxis, neurogenic | Widespread vasodilation → relative hypovolemia → maldistribution of flow |
| Obstructive | Mechanical obstruction to flow | Tamponade, tension pneumothorax, massive PE | Impaired ventricular filling or outflow → decreased CO |
Response to Hemorrhage
Acute blood loss triggers a coordinated compensatory response: (1) Baroreceptor reflex (seconds): decreased carotid sinus/aortic arch stretch → decreased parasympathetic, increased sympathetic tone → tachycardia, vasoconstriction, increased contractility. (2) Chemoreceptor activation (minutes): decreased O2 delivery stimulates peripheral chemoreceptors → increased ventilation. (3) RAAS activation (minutes-hours): decreased renal perfusion → renin → angiotensin II → vasoconstriction + aldosterone → Na+/water retention. (4) ADH release (minutes): hypovolemia is a potent non-osmotic stimulus. (5) Fluid shifts (hours): decreased capillary hydrostatic pressure → interstitial fluid moves into the vascular space (transcapillary refill). (6) Erythropoietin (days): renal hypoxia stimulates EPO production → increased RBC production.
Aging & Physiologic Changes
| System | Change with Aging | Clinical Consequence |
|---|---|---|
| Cardiovascular | Increased arterial stiffness, decreased maximum HR, LV hypertrophy, decreased baroreceptor sensitivity | Isolated systolic hypertension, orthostatic hypotension, decreased exercise capacity |
| Respiratory | Decreased chest wall compliance, increased residual volume, decreased FEV1/FVC, decreased PaO2 | Decreased respiratory reserve, increased A-a gradient (normal aging: A-a = 2.5 + 0.21 x age) |
| Renal | Decreased GFR (~1 mL/min/year after age 40), decreased concentrating ability, decreased renin/aldosterone | Increased drug half-lives, impaired Na+/water handling, risk of hyponatremia |
| Endocrine | Decreased T-cell function, increased insulin resistance, decreased GH/IGF-1, decreased estrogen/testosterone | Type 2 diabetes risk, osteoporosis, sarcopenia, immune senescence |
| Neurologic | Decreased brain mass, decreased neurotransmitter synthesis, slower conduction velocity | Slower reflexes, decreased memory, increased fall risk |
Cardiovascular Changes in Pregnancy
| Parameter | Change | Mechanism |
|---|---|---|
| Blood volume | Increases 40-50% | RAAS activation, estrogen-mediated fluid retention |
| Cardiac output | Increases 30-50% | Increased HR + increased SV |
| Heart rate | Increases 15-20 bpm | Compensatory for increased CO demand |
| SVR | Decreases | Progesterone-mediated vasodilation, low-resistance placental circuit |
| Blood pressure | Decreases in 2nd trimester, normalizes by term | Decreased SVR outpaces increased CO initially |
| Plasma volume vs RBC mass | Plasma increases more → dilutional anemia | "Physiologic anemia of pregnancy" (Hb ~11 g/dL normal) |
Temperature Regulation
The hypothalamus (preoptic/anterior region) is the thermoregulatory center. Responses to heat: vasodilation (increased skin blood flow via sympathetic withdrawal), sweating (cholinergic sympathetic fibers), behavioral changes. Responses to cold: vasoconstriction (alpha-1 in skin), shivering (involuntary muscle contraction generating heat), non-shivering thermogenesis (UCP1 in brown fat — important in neonates), piloerection. Fever results from pyrogens (IL-1, IL-6, TNF, PGE2) raising the hypothalamic set point. Antipyretics (acetaminophen, NSAIDs) work by inhibiting COX → reducing PGE2 synthesis in the hypothalamus.
High-Altitude Physiology
At altitude, barometric pressure decreases → lower inspired PO2 → hypoxemia. Acute acclimatization (hours-days): peripheral chemoreceptors detect hypoxemia → hyperventilation → respiratory alkalosis → renal compensation (bicarbonaturia via decreased HCO3- reabsorption). Acetazolamide (CA inhibitor) accelerates renal compensation by causing metabolic acidosis, which drives further hyperventilation. Chronic acclimatization (weeks): increased EPO → polycythemia (increased O2-carrying capacity); increased 2,3-DPG → right-shifted O2-Hb curve (enhanced tissue O2 delivery); pulmonary vascular remodeling (increased pulmonary artery pressure). Acute mountain sickness: headache, nausea, fatigue at >2500 m. HAPE (high-altitude pulmonary edema): noncardiogenic pulmonary edema from exaggerated HPV; treatment = descent + supplemental O2 + nifedipine. HACE (high-altitude cerebral edema): vasogenic brain edema causing ataxia, altered mentation; treatment = immediate descent + dexamethasone.
Diving Physiology & Gas Laws
| Gas Law | Statement | Clinical Application |
|---|---|---|
| Boyle's Law | P1V1 = P2V2 (at constant T) | Descent: gas spaces compress (ear squeeze); Ascent: gas spaces expand (pneumothorax risk, barotrauma) |
| Dalton's Law | Ptotal = sum of partial pressures | At depth, increased N2 partial pressure → nitrogen narcosis (at ~30 m); O2 toxicity at very high PO2 (seizures) |
| Henry's Law | Gas dissolved in liquid is proportional to partial pressure | At depth, more N2 dissolves in tissues; rapid ascent → N2 comes out of solution as bubbles → decompression sickness ("the bends") |
27 High-Yield Review & Key Equations
Essential Equations
| Equation | Formula | Application |
|---|---|---|
| Fick Principle | CO = VO2 / (CaO2 - CvO2) | Gold standard for cardiac output measurement |
| MAP | CO x SVR; also DBP + 1/3(PP) | Organ perfusion pressure |
| Starling Equation | Q = Kf[(Pc - Pi) - sigma(pic - pii)] | Capillary fluid exchange, edema formation |
| Alveolar Gas Equation | PAO2 = FiO2(Patm - PH2O) - (PaCO2/R) | Calculate expected alveolar PO2, A-a gradient |
| Henderson-Hasselbalch | pH = 6.1 + log([HCO3-] / 0.03 x PaCO2) | Acid-base analysis |
| GFR (Clearance) | Cx = (Ux x V) / Px | Renal function assessment |
| Filtration Fraction | FF = GFR / RPF | Normal ~20%; increases with efferent constriction |
| Nernst Equation | E = (61.5/z) x log([ion]out/[ion]in) | Equilibrium potential for single ion |
| Winter's Formula | Expected PaCO2 = 1.5(HCO3-) + 8 +/- 2 | Expected respiratory compensation in metabolic acidosis |
| Laplace Law (alveolus) | P = 2T/r | Surfactant function, why small alveoli tend to collapse |
| Poiseuille's Law | R proportional to 1/r4 | Airway and vascular resistance |
| O2 Content | CaO2 = (1.34 x Hb x SaO2) + (0.003 x PaO2) | Total oxygen carrying capacity of blood |
High-Yield Physiology Concepts by System
Cardiovascular: Frank-Starling law, PV loops, Fick principle, baroreceptor reflex, hemodynamic profiles of shock, cardiac action potential phases, S2 splitting.
Respiratory: V/Q matching (shunt vs dead space), alveolar gas equation, A-a gradient, O2-Hb dissociation curve shifts, compliance/surfactant, lung volumes (FRC, RV).
Renal: GFR/clearance equations, nephron segment transporters and diuretic targets, RAAS, countercurrent mechanism, ADH physiology, free water clearance.
Acid-Base: Henderson-Hasselbalch, compensation rules (Winter's formula), anion gap, delta-delta analysis, RTA classification.
GI: Acid secretion regulation, CCK/secretin functions, bile salt enterohepatic circulation, absorption sites (iron = duodenum, B12 = terminal ileum).
Neuro: Action potential phases, neurotransmitter systems, UMN vs LMN signs, DCML vs spinothalamic tracts, ANS receptor pharmacology.
Endocrine: HPA/HPT/HPG axes, second messenger systems, PTH vs vitamin D, thyroid physiology, adrenal zones.
Muscle: Excitation-contraction coupling, skeletal vs cardiac vs smooth muscle differences, malignant hyperthermia.
Physiologic Responses — Summary of Key Reflexes
| Reflex/Response | Stimulus | Effector | Result |
|---|---|---|---|
| Baroreceptor reflex | Change in arterial BP (carotid sinus CN IX, aortic arch CN X) | Autonomic NS → heart, vessels | Buffers acute BP changes (increased BP → decreased HR/SVR; decreased BP → increased HR/SVR) |
| Chemoreceptor reflex | Decreased PaO2 (< 60), increased PaCO2, decreased pH | Respiratory centers → ventilatory muscles | Increased ventilation |
| Cushing reflex | Increased ICP compressing brainstem | Sympathetic surge → vagal response | Hypertension + bradycardia + irregular breathing (Cushing triad — ominous sign of herniation) |
| Bainbridge reflex | Increased venous return (atrial stretch) | Vagal withdrawal + sympathetic activation | Increased HR (prevents atrial congestion) |
| Bezold-Jarisch reflex | LV mechanoreceptor activation (stretch in underfilled ventricle) | Increased vagal tone | Bradycardia + hypotension + vasodilation (vasovagal syncope mechanism) |
| Hering-Breuer reflex | Lung overinflation (pulmonary stretch receptors) | Vagus nerve → respiratory center | Terminates inspiration (prevents overinflation) |
| Tubuloglomerular feedback | Increased NaCl at macula densa | Afferent arteriole (adenosine-mediated constriction) | Decreased GFR (protects against excessive filtration) |
Commonly Tested Comparisons
| Concept A | Concept B | Key Distinction |
|---|---|---|
| Shunt (V/Q = 0) | Dead space (V/Q = infinity) | Shunt does NOT correct with O2; dead space does |
| Compliance | Elastance | Compliance = 1/elastance; high compliance = easily stretched |
| UMN lesion | LMN lesion | UMN: spastic, hyperreflexia, Babinski+; LMN: flaccid, areflexia, fasciculations |
| Central DI | Nephrogenic DI | Central: responds to desmopressin; Nephrogenic: does not |
| Metabolic acidosis compensation | Metabolic alkalosis compensation | Acidosis compensation is more effective (hypoxia limits hypoventilation in alkalosis) |
| Type I RTA (distal) | Type II RTA (proximal) | Type I: cannot secrete H+ (urine pH > 5.5); Type II: cannot reabsorb HCO3- (urine pH initially > 5.5 then < 5.5) |
| PTH effect on Ca2+ | PTH effect on PO43- | PTH raises Ca2+ and LOWERS PO43- (phosphaturic) |
| Bohr effect | Haldane effect | Bohr: CO2/H+ reduces O2 affinity; Haldane: deoxyHb carries more CO2 |
| SIADH | Cerebral salt wasting | Both: hyponatremia; SIADH = euvolemic; CSW = hypovolemic |
| Conn syndrome | Addison disease | Conn: hypertension + hypokalemia + alkalosis; Addison: hypotension + hyperkalemia + acidosis |
Rapid-Fire Physiology Facts
| Fact | Detail |
|---|---|
| Fastest conduction in the heart | Purkinje fibers (~4 m/s) |
| Slowest conduction in the heart | AV node (~0.05 m/s) — intentional delay for atrial filling |
| Highest O2 extraction | Heart (~70-75%); coronary sinus PO2 is lowest in the body |
| Largest blood reservoir | Venous system (~60-70% of blood volume) |
| Site of most airway resistance | Medium-sized bronchi (not smallest airways) |
| Only artery carrying deoxygenated blood | Pulmonary artery |
| Only vein carrying oxygenated blood | Pulmonary vein |
| Organ that uses ketones preferentially in starvation | Brain (after 3-4 days of fasting; normally uses only glucose) |
| RBC fuel source | Glucose only (no mitochondria — anaerobic glycolysis) |
| Rate-limiting enzyme of cholesterol synthesis | HMG-CoA reductase (target of statins) |
| Most common buffer in urine | NH3/NH4+ (from glutamine metabolism in PCT) |
| Hormone that increases in ALL types of shock | ADH (vasopressin) — released by hypovolemia and hypotension |
Physiology is never tested in isolation. The board exam and clinical practice require integrating across systems: a patient with heart failure has cardiac dysfunction (decreased CO), pulmonary congestion (Starling forces), renal compensation (RAAS activation), acid-base disturbances, and electrolyte shifts — all connected by the same physiologic principles. A patient with cirrhosis demonstrates splanchnic vasodilation (decreased effective circulating volume), RAAS activation (sodium and water retention), decreased albumin synthesis (decreased oncotic pressure → ascites and edema), impaired bile salt production (fat malabsorption), and portosystemic shunting (hepatic encephalopathy from bypassed ammonia metabolism). Mastering these cross-system connections is the key to clinical excellence.