Hematology / Oncology
Every blood disorder, malignancy, coagulation pathway, chemotherapy regimen, transfusion protocol, and oncologic emergency in one place.
01 Hematopoiesis & Blood Cell Biology
All blood cells originate from a common pluripotent hematopoietic stem cell (HSC) residing in the bone marrow. HSCs are capable of self-renewal and differentiation into every mature blood lineage. Understanding normal hematopoiesis is essential for interpreting cytopenias, marrow failure syndromes, and hematologic malignancies.
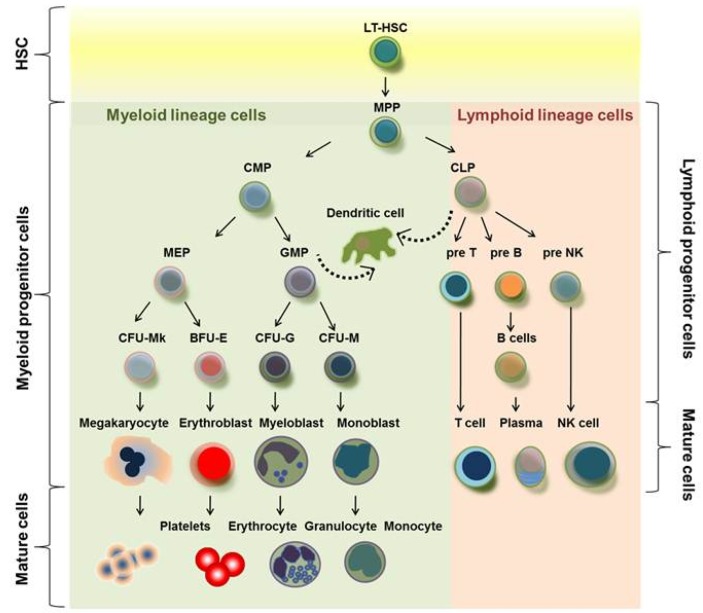
Hematopoietic Hierarchy
The HSC gives rise to two major progenitor lineages: the common myeloid progenitor (CMP) and the common lymphoid progenitor (CLP). The CMP differentiates into erythrocytes, platelets (via megakaryocytes), granulocytes (neutrophils, eosinophils, basophils), and monocytes/macrophages. The CLP gives rise to B lymphocytes, T lymphocytes, and natural killer (NK) cells. Each step is driven by specific growth factors and transcription factors.
Key Growth Factors
| Growth Factor | Target Lineage | Clinical Use |
|---|---|---|
| Erythropoietin (EPO) | Erythroid progenitors | Anemia of CKD, MDS; produced by renal peritubular cells in response to hypoxia |
| Thrombopoietin (TPO) | Megakaryocytes | Romiplostim, eltrombopag for ITP; produced primarily by the liver |
| G-CSF (filgrastim) | Neutrophil precursors | Chemotherapy-induced neutropenia, stem cell mobilization |
| GM-CSF (sargramostim) | Granulocytes & monocytes | Post-transplant marrow recovery |
| SCF (stem cell factor) | HSCs, mast cells | Experimental; c-KIT ligand |
| IL-3 | Multi-lineage early progenitors | Broad stimulatory; rarely used clinically |
Bone Marrow Microenvironment
The marrow niche consists of osteoblastic (endosteal) and vascular (sinusoidal) niches that regulate HSC quiescence, self-renewal, and mobilization. Stromal cells, extracellular matrix, and cytokines (CXCL12/SDF-1 and its receptor CXCR4) anchor HSCs in the niche. Disruption of this microenvironment contributes to myelofibrosis, aplastic anemia, and leukemogenesis. In adults, active (red) marrow is concentrated in the axial skeleton (vertebrae, pelvis, sternum, ribs, proximal femur/humerus), while yellow (fatty) marrow predominates in the distal long bones.
Normal Blood Cell Lifespans
| Cell Type | Normal Count | Lifespan | Key Function |
|---|---|---|---|
| Red blood cell | 4.5–5.5 × 1012/L | ~120 days | Oxygen transport via hemoglobin |
| Platelet | 150–400 × 109/L | 8–10 days | Primary hemostasis, plug formation |
| Neutrophil | 2.0–7.0 × 109/L | 6–8 hours (blood) | Bacterial defense, phagocytosis |
| Lymphocyte | 1.0–4.0 × 109/L | Weeks to years (memory) | Adaptive immunity (B-cell, T-cell, NK) |
| Monocyte | 0.2–0.8 × 109/L | 1–3 days (blood) → months (tissue macrophage) | Phagocytosis, antigen presentation |
| Eosinophil | 0.04–0.4 × 109/L | 8–12 hours (blood) | Parasitic defense, allergy |
02 The Hematologic Assessment
Complete Blood Count (CBC) Interpretation
| Parameter | Normal Range | Clinical Significance When Abnormal |
|---|---|---|
| Hemoglobin (Hb) | M: 13.5–17.5 g/dL; F: 12–16 g/dL | Low = anemia; High = polycythemia |
| Hematocrit (Hct) | M: 41–53%; F: 36–46% | Approximately 3× Hb value |
| MCV | 80–100 fL | <80 = microcytic; >100 = macrocytic |
| MCH | 27–33 pg | Low in iron deficiency and thalassemia |
| MCHC | 32–36 g/dL | Elevated in spherocytosis; low in iron deficiency |
| RDW | 11.5–14.5% | Elevated = anisocytosis (mixed cell sizes); helps distinguish IDA (high RDW) from thalassemia trait (normal RDW) |
| WBC | 4.5–11.0 × 109/L | Leukocytosis or leukopenia; always check differential |
| Platelet count | 150–400 × 109/L | <150 = thrombocytopenia; >400 = thrombocytosis |
| Reticulocyte count | 0.5–2.5% (absolute 25–125 × 109/L) | Corrected reticulocyte count = retic % × (patient Hct / normal Hct); reticulocyte production index (RPI) further corrects for marrow release |
Peripheral Blood Smear Findings
| Finding | Description | Associated Conditions |
|---|---|---|
| Target cells | Bull's-eye appearance | Thalassemia, liver disease, hemoglobin C, post-splenectomy |
| Schistocytes | Fragmented RBCs (helmet cells) | TTP, HUS, DIC, HELLP, mechanical valve hemolysis |
| Spherocytes | Small, dense, no central pallor | Hereditary spherocytosis, autoimmune hemolytic anemia (AIHA) |
| Sickle cells (drepanocytes) | Crescent-shaped RBCs | Sickle cell disease |
| Howell-Jolly bodies | Nuclear remnants in RBCs | Asplenia or hyposplenia |
| Teardrop cells (dacrocytes) | Teardrop-shaped RBCs | Myelofibrosis, marrow infiltration |
| Rouleaux formation | Stacked coins appearance | Multiple myeloma, inflammation (high ESR) |
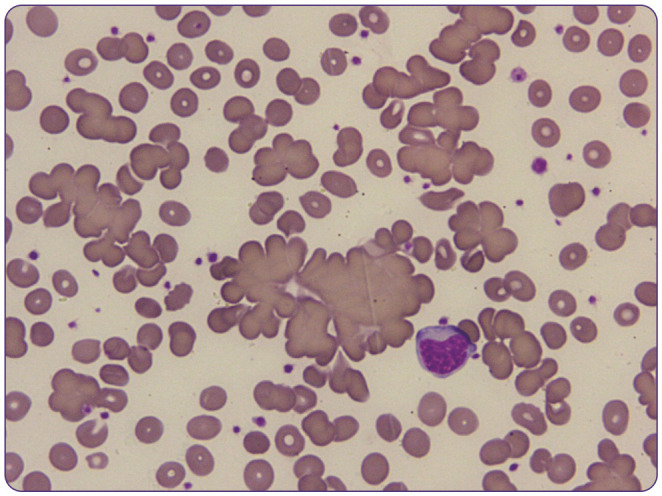
| Hypersegmented neutrophils | ≥5 lobes (or ≥1 with 6+ lobes) | Megaloblastic anemia (B12/folate deficiency) |
| Auer rods | Needle-like cytoplasmic inclusions | AML (especially APL — M3) |
| Smudge cells | Fragile lymphocytes disrupted on smear | CLL |
Hemolysis Workup
| Test | Expected in Hemolysis | Notes |
|---|---|---|
| LDH | Elevated | Released from lysed RBCs; nonspecific |
| Haptoglobin | Low / undetectable | Binds free hemoglobin; consumed during intravascular hemolysis |
| Indirect bilirubin | Elevated | From hemoglobin catabolism |
| Reticulocyte count | Elevated | Appropriate marrow response |
| Direct Coombs (DAT) | Positive in immune-mediated | Detects IgG or complement (C3d) on RBC surface |
| Peripheral smear | Schistocytes, spherocytes | Morphology guides differential diagnosis |
Coagulation Studies
| Test | Pathway | Normal | Prolonged In |
|---|---|---|---|
| PT / INR | Extrinsic (VII) + common (X, V, II, fibrinogen) | 11–13.5 sec / INR 0.8–1.2 | Warfarin, liver disease, vitamin K deficiency, DIC |
| aPTT | Intrinsic (XII, XI, IX, VIII) + common | 25–35 sec | Heparin, hemophilia A/B, lupus anticoagulant, DIC |
| Thrombin time (TT) | Fibrinogen → fibrin | 14–19 sec | Heparin contamination, fibrinogen disorders, dabigatran |
| Fibrinogen | Common pathway substrate | 200–400 mg/dL | Low in DIC, liver failure; acute phase reactant (may be elevated) |
| D-dimer | Fibrin degradation product | <500 ng/mL | Elevated in DVT/PE, DIC, post-surgical, infection (sensitive but not specific) |
| Mixing study | Distinguishes factor deficiency vs inhibitor | — | Corrects → factor deficiency; does not correct → inhibitor (e.g., lupus anticoagulant, factor inhibitor) |
03 Key Terminology & Abbreviations
04 Iron Deficiency Anemia
Iron deficiency anemia (IDA) is the most common cause of anemia worldwide, affecting an estimated 1–2 billion people. Iron is essential for hemoglobin synthesis, and depletion progresses through stages: iron depletion (low ferritin) → iron-deficient erythropoiesis (low transferrin saturation) → frank IDA (microcytic, hypochromic anemia).
Etiology
| Mechanism | Common Causes |
|---|---|
| Blood loss (most common in adults) | GI bleeding (peptic ulcer, colon cancer, angiodysplasia), menstruation, surgical |
| Decreased absorption | Celiac disease, gastric bypass, H. pylori, PPI use, inflammatory bowel disease |
| Increased demand | Pregnancy, lactation, rapid growth in children/adolescents |
| Inadequate intake | Vegan/vegetarian diet without supplementation, food insecurity |
Iron Studies Interpretation
| Parameter | IDA | Anemia of Chronic Disease | Thalassemia Trait | Sideroblastic |
|---|---|---|---|---|
| Serum iron | ↓ | ↓ | Normal/↑ | ↑ |
| TIBC | ↑ | ↓ | Normal | Normal |
| Transferrin sat | ↓ (<20%) | ↓ | Normal | ↑ |
| Ferritin | ↓ (<30 ng/mL) | Normal/↑ | Normal | ↑ |
| MCV | ↓ | Normal/↓ | ↓ | Normal/↓ |
| RDW | ↑ | Normal | Normal | ↑ |
Management
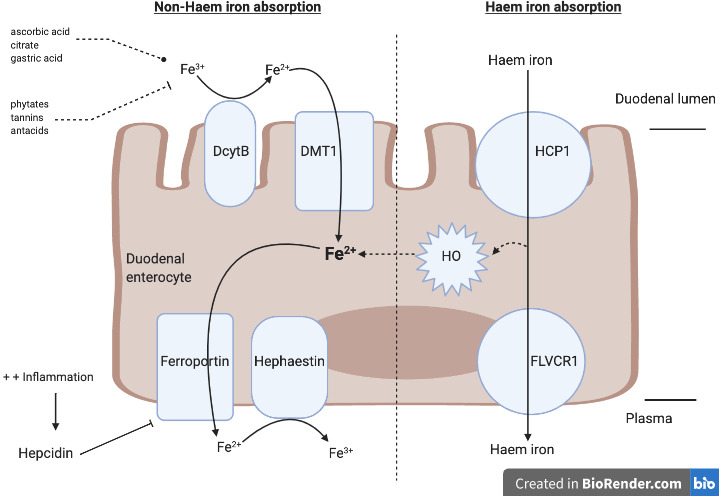
Oral iron: Ferrous sulfate 325 mg (65 mg elemental iron) PO daily to TID on an empty stomach with vitamin C to enhance absorption. Expect hemoglobin rise of ~1 g/dL every 2–3 weeks. Continue for 3–6 months after Hb normalizes to replete stores. Common side effects include nausea, constipation, and dark stools.
IV iron: Indicated when oral iron fails, is not tolerated, or absorption is impaired (celiac, gastric bypass). Options include iron sucrose (Venofer), ferric carboxymaltose (Injectafer — allows single high-dose infusion of 750 mg), and ferumoxytol (Feraheme). Low dose iron dextran is rarely used due to anaphylaxis risk.
All adults with new-onset IDA require investigation for occult GI blood loss. Men and postmenopausal women should be evaluated with upper and lower endoscopy unless an obvious non-GI source is identified. Do not attribute IDA to menstruation without confirming menstrual history.
05 Megaloblastic Anemia
Megaloblastic anemias are characterized by impaired DNA synthesis leading to nuclear-cytoplasmic dyssynchrony and macrocytosis (MCV >100 fL). The two principal causes are vitamin B12 (cobalamin) and folate deficiency.
Vitamin B12 Deficiency
| Feature | Details |
|---|---|
| Causes | Pernicious anemia (autoimmune destruction of parietal cells — most common in developed countries), gastrectomy/bypass, ileal resection/Crohn's, strict veganism, metformin, nitrous oxide exposure |
| Hematologic findings | Macrocytic anemia, hypersegmented neutrophils, pancytopenia in severe cases; elevated LDH and indirect bilirubin (ineffective erythropoiesis) |
| Neurologic manifestations | Subacute combined degeneration: demyelination of dorsal columns (loss of proprioception, vibration) and lateral corticospinal tracts (spasticity, weakness); peripheral neuropathy, cognitive decline, psychosis |
| Diagnosis | Low serum B12 (<200 pg/mL); elevated methylmalonic acid (MMA) and homocysteine; anti-intrinsic factor antibodies (specific for pernicious anemia); anti-parietal cell antibodies (sensitive but less specific) |
| Treatment | IM cyanocobalamin 1000 mcg daily × 7 days, then weekly × 4 weeks, then monthly for life (if pernicious anemia); high-dose oral B12 (1000–2000 mcg/day) acceptable if absorption issue is not severe |
Folate Deficiency
| Feature | Details |
|---|---|
| Causes | Alcoholism (most common), poor dietary intake, malabsorption (celiac), increased demand (pregnancy, hemolytic anemias), medications (methotrexate, trimethoprim, phenytoin) |
| Lab findings | Low serum folate (<3 ng/mL), elevated homocysteine, normal MMA (distinguishes from B12 deficiency) |
| Neurologic | No neurologic findings (unlike B12 deficiency) |
| Treatment | Folic acid 1–5 mg PO daily; correct underlying cause |
Always check B12 before treating with folate alone. Folate supplementation can correct the anemia of B12 deficiency but will NOT reverse or prevent neurologic damage, which may become irreversible.
06 Hemolytic Anemias
Hemolytic anemias result from premature RBC destruction (lifespan <120 days). They are classified as intrinsic (defect within the RBC — membrane, enzyme, or hemoglobin) or extrinsic (external factors acting on normal RBCs — immune, mechanical, infectious). Hemolysis may be intravascular (within the bloodstream) or extravascular (splenic/hepatic macrophages).
Classification
| Category | Examples | Key Features |
|---|---|---|
| Membrane defects | Hereditary spherocytosis, hereditary elliptocytosis, PNH | Spherocytes on smear (HS); osmotic fragility test positive; PNH — flow cytometry for CD55/CD59 loss |
| Enzyme defects | G6PD deficiency, pyruvate kinase deficiency | G6PD: X-linked; episodic hemolysis triggered by oxidative stress (fava beans, sulfa drugs, infections); Heinz bodies and bite cells on smear |
| Hemoglobinopathies | Sickle cell disease, thalassemias | Covered in Sections 07 and 08 |
| Immune-mediated | Warm AIHA, cold agglutinin disease, drug-induced | DAT positive; warm AIHA (IgG, extravascular, treat with steroids/rituximab); cold agglutinin (IgM/complement, intravascular, avoid cold) |
| Microangiopathic | TTP, HUS, DIC, HELLP | Schistocytes on smear; thrombocytopenia; elevated LDH; DAT negative |
| Mechanical | Prosthetic heart valve, march hemoglobinuria | Schistocytes; LDH elevated; intravascular hemolysis |
| Infectious | Malaria, Babesia, Clostridium perfringens | Direct RBC parasitization or toxin-mediated lysis |
Direct Antiglobulin Test (Coombs Test)
The direct Coombs test (DAT) detects antibodies or complement bound to the patient's RBC surface. A positive DAT with IgG suggests warm AIHA (extravascular hemolysis mediated by splenic macrophages). A positive DAT with C3d suggests cold agglutinin disease (IgM activates complement at cooler temperatures). The indirect Coombs test detects free antibodies in the patient's serum (used in blood bank crossmatching and prenatal testing).
Paroxysmal Nocturnal Hemoglobinuria (PNH)
PNH is caused by an acquired somatic mutation in the PIGA gene in HSCs, leading to loss of GPI-anchored proteins (CD55 and CD59) that normally protect RBCs from complement-mediated lysis. Triad: hemolytic anemia + venous thrombosis (especially hepatic/Budd-Chiari, cerebral) + cytopenias. Diagnosis: flow cytometry showing loss of CD55/CD59. Treatment: eculizumab (anti-C5 monoclonal antibody) dramatically reduces hemolysis and thrombotic risk; ravulizumab (longer half-life C5 inhibitor) is an alternative.
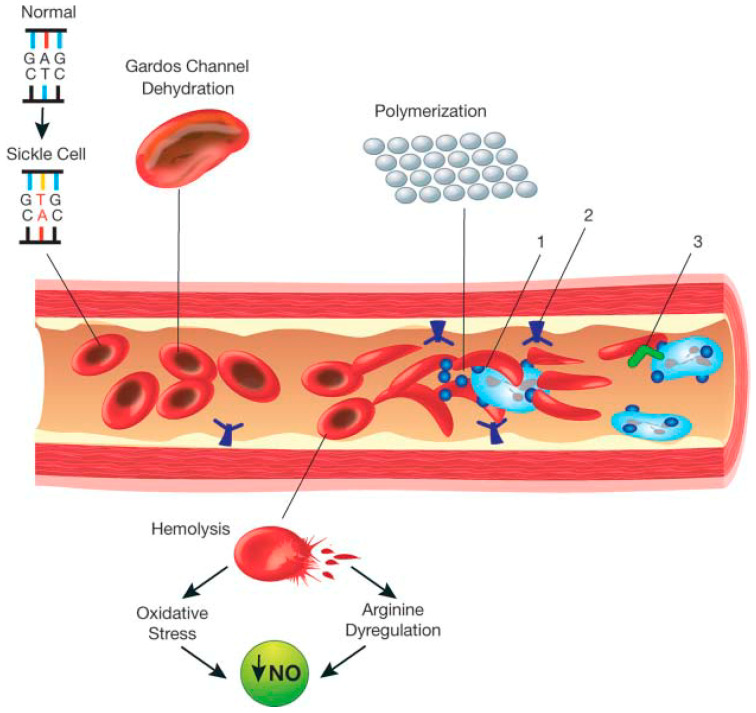
07 Sickle Cell Disease
Sickle cell disease (SCD) is an autosomal recessive hemoglobinopathy caused by a point mutation in the beta-globin gene (Glu → Val at position 6), producing hemoglobin S (HbS). Under deoxygenated conditions, HbS polymerizes, causing RBCs to sickle. These rigid cells occlude the microvasculature, causing ischemia, hemolysis, and progressive organ damage. Prevalence is highest in populations from sub-Saharan Africa, the Mediterranean, Middle East, and India (heterozygous advantage against falciparum malaria).
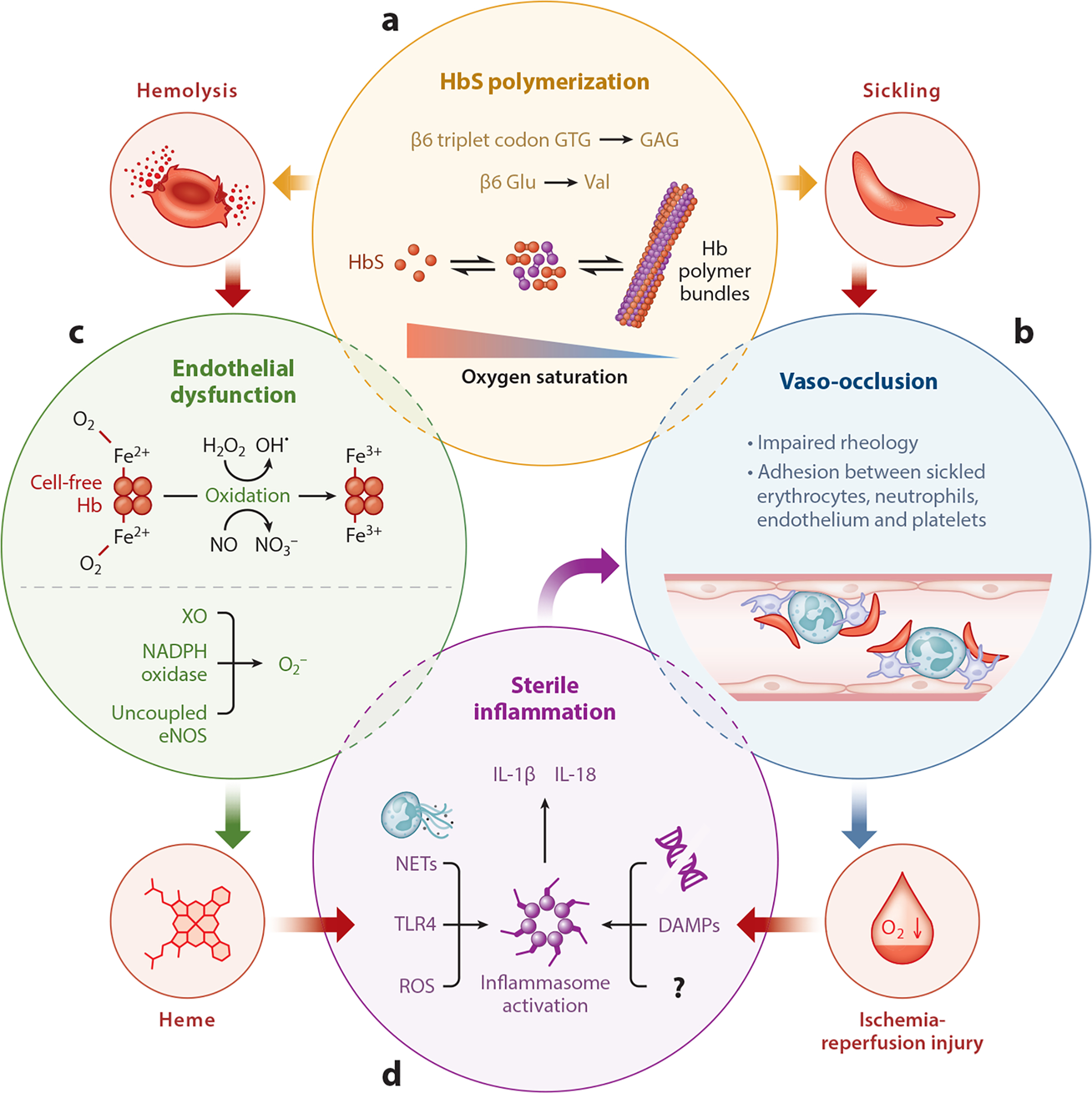
Genotypes
| Genotype | Severity | HbS % | Notes |
|---|---|---|---|
| HbSS (sickle cell anemia) | Severe | ~80–90% | Most common and most severe form |
| HbSC | Moderate | ~50% | Milder anemia; higher risk of proliferative retinopathy and avascular necrosis |
| HbS/β0-thalassemia | Severe | ~80–90% | Clinically similar to HbSS |
| HbS/β+-thalassemia | Mild-Moderate | ~60–75% | Some normal HbA present |
| Sickle cell trait (HbAS) | Generally benign | ~40% | Rarely symptomatic; risk of renal medullary carcinoma, hematuria, splenic infarction at altitude |
Acute Complications
| Complication | Features | Management |
|---|---|---|
| Vaso-occlusive crisis (VOC) | Severe pain in bones, chest, abdomen; most common reason for ED visits | IV fluids, multimodal analgesia (opioids + NSAIDs + acetaminophen), incentive spirometry; avoid under-treating pain |
| Acute chest syndrome (ACS) | New pulmonary infiltrate + fever/respiratory symptoms; leading cause of death in adults with SCD | Antibiotics (cephalosporin + macrolide), supplemental O2, simple or exchange transfusion (target HbS <30%), bronchodilators, incentive spirometry |
| Stroke | Ischemic (children) or hemorrhagic (adults); 11% lifetime risk | Exchange transfusion acutely; chronic transfusion program (target HbS <30%); transcranial Doppler screening in children 2–16 years |
| Aplastic crisis | Sudden drop in Hb with low reticulocyte count; parvovirus B19 | Supportive care, transfusion; usually self-limited (5–10 days) |
| Splenic sequestration | Acute splenomegaly + severe anemia (blood pooling in spleen) | Emergent transfusion; recurrent episodes → splenectomy |
| Priapism | Prolonged painful erection >4 hours | Aspiration/irrigation, phenylephrine injection; recurrent → hydroxyurea |
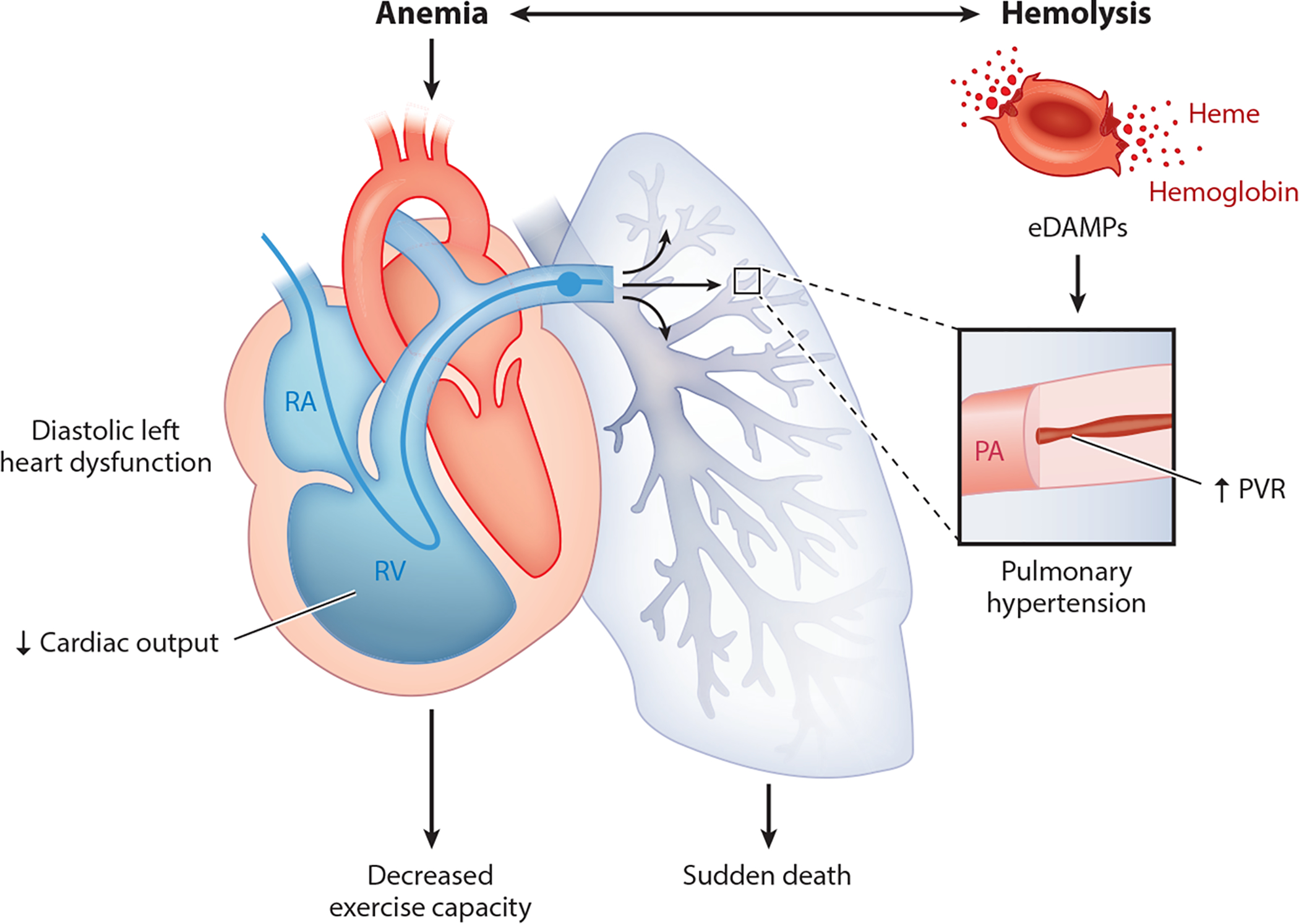
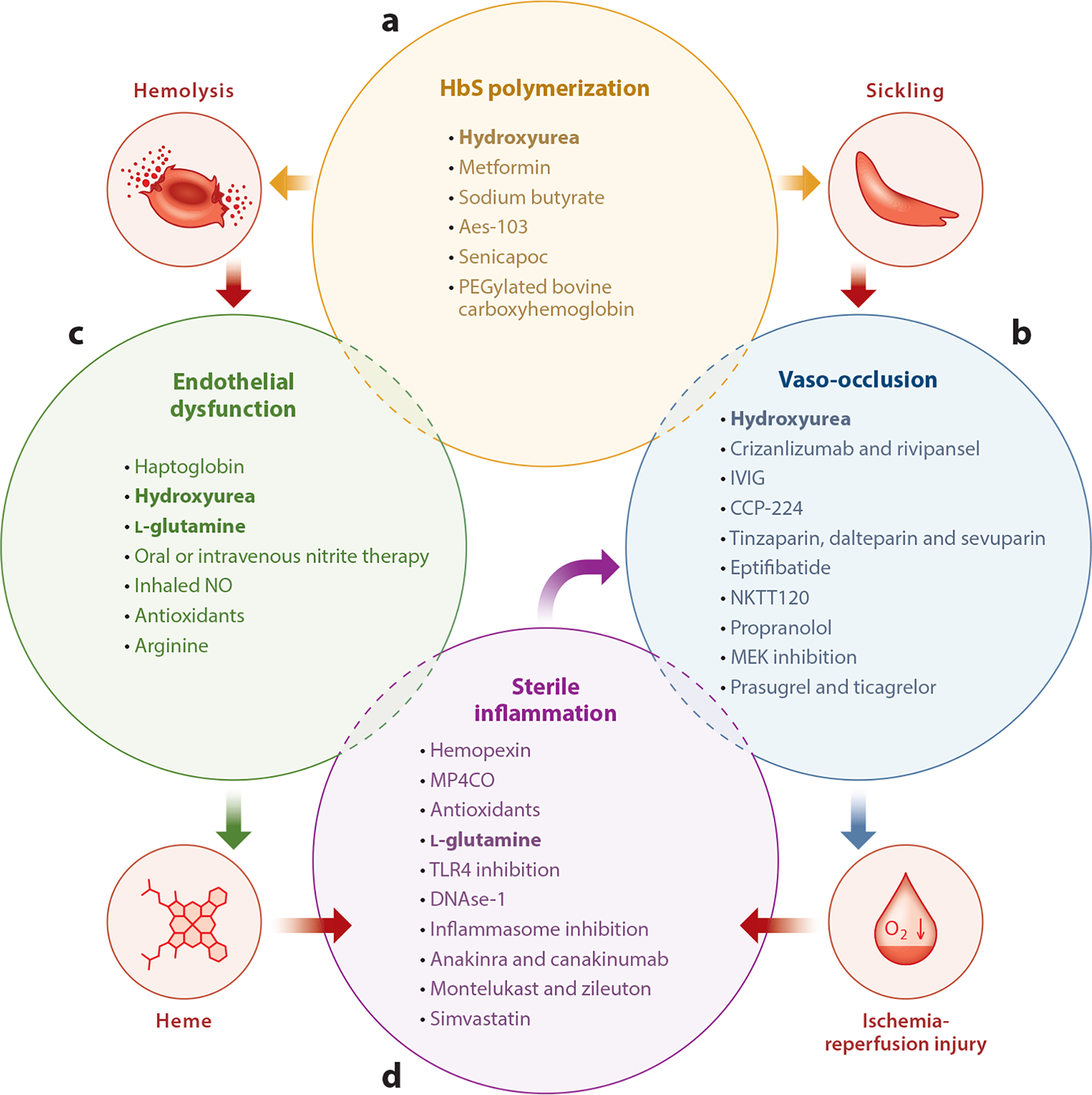
Disease-Modifying Therapies
| Agent | Mechanism | Key Points |
|---|---|---|
| Hydroxyurea | Increases fetal hemoglobin (HbF), which inhibits HbS polymerization | First-line disease-modifying therapy; reduces VOC, ACS, transfusions, and mortality; monitor CBC for myelosuppression |
| L-glutamine (Endari) | Reduces oxidative stress in sickle RBCs | FDA-approved 2017; reduces VOC frequency; can be added to hydroxyurea |
| Voxelotor (Oxbryta) | HbS polymerization inhibitor (increases Hb-oxygen affinity) | Increases Hb, reduces hemolysis markers; FDA-approved 2019 |
| Crizanlizumab (Adakveo) | Anti-P-selectin antibody (reduces vaso-occlusion) | Reduces annual VOC rate; IV monthly |
| Allogeneic stem cell transplant | Curative | Best outcomes with matched sibling donor in children; only established cure (until gene therapy) |
| Gene therapy (lovotibeglogene, exagamglogene) | Gene addition or CRISPR gene editing to increase HbF | FDA-approved 2023 (Casgevy — CRISPR; Lyfgenia — lentiviral); potentially curative |
ACS can rapidly progress to respiratory failure and death. Any sickle cell patient with new infiltrate on CXR + fever, chest pain, or hypoxia has ACS until proven otherwise. Start empiric antibiotics, oxygen, and arrange exchange transfusion immediately if Hb is near baseline or worsening.
08 Thalassemias
The thalassemias are inherited hemoglobinopathies characterized by decreased or absent production of one or more globin chains. This leads to ineffective erythropoiesis, hemolysis, and microcytic anemia. They are classified as alpha-thalassemia (reduced alpha-globin) or beta-thalassemia (reduced beta-globin).
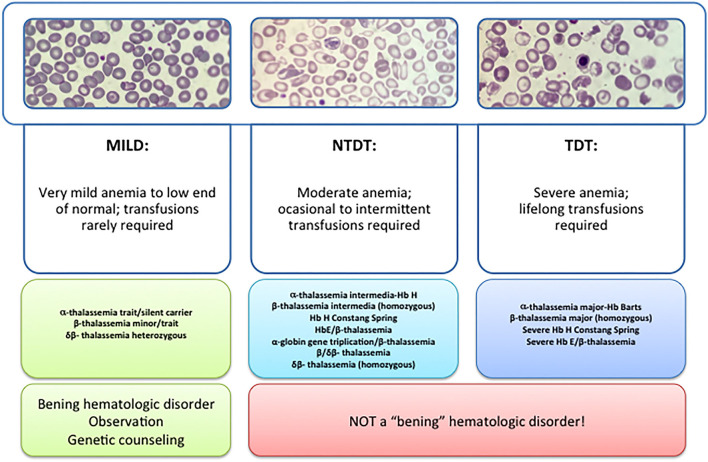
Alpha-Thalassemia
| Gene Deletions | Condition | Clinical Features | Hemoglobin Pattern |
|---|---|---|---|
| 1 (αα/α–) | Silent carrier | Normal CBC or minimal microcytosis | Normal |
| 2 (α–/α– or αα/––) | Alpha-thal trait | Mild microcytic anemia; can mimic IDA | Normal (no HbH); diagnosis by genetic testing |
| 3 (α–/––) | HbH disease | Moderate hemolytic anemia, splenomegaly | HbH (beta-4 tetramers) on electrophoresis |
| 4 (––/––) | Hb Bart's hydrops fetalis | Incompatible with life; in utero death | Hb Bart's (gamma-4 tetramers); no functional hemoglobin |
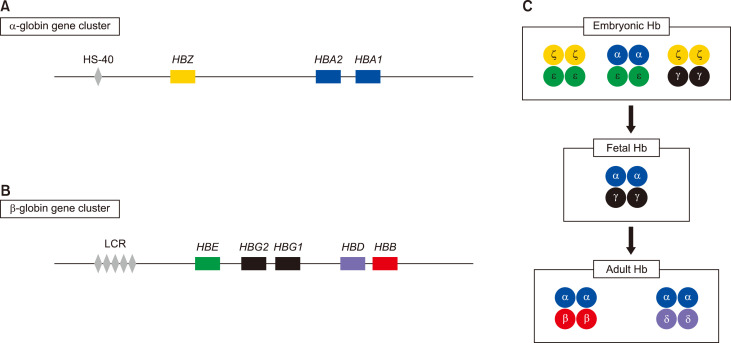
Beta-Thalassemia
| Type | Genotype | Clinical Features | Hemoglobin Electrophoresis |
|---|---|---|---|
| Beta-thal minor (trait) | β/β+ or β/β0 | Mild microcytic anemia; normal RDW; target cells; often mistaken for IDA | Elevated HbA2 (>3.5%) — diagnostic |
| Beta-thal intermedia | β+/β+ | Moderate anemia; variable transfusion need; iron overload from increased absorption | Elevated HbA2 and HbF; reduced HbA |
| Beta-thal major (Cooley's) | β0/β0 | Severe anemia presenting at 6–12 months; transfusion-dependent; hepatosplenomegaly, skeletal deformities (chipmunk facies, hair-on-end skull) | HbF 60–90%; absent or minimal HbA; elevated HbA2 |
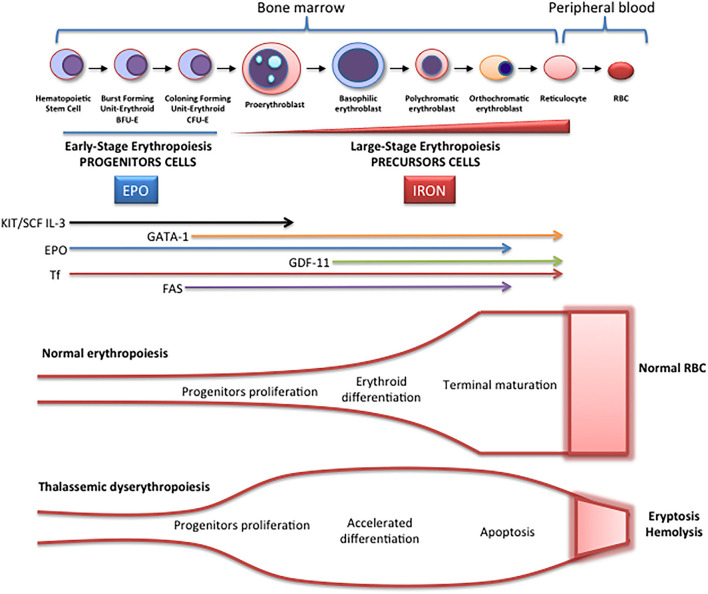
Management of Thalassemia Major
Chronic transfusion: Regular RBC transfusions every 2–4 weeks to maintain Hb 9–10.5 g/dL. This suppresses ineffective erythropoiesis and prevents skeletal deformities. Iron chelation is mandatory to prevent transfusional iron overload (hemochromatosis). Options: deferoxamine (SC/IV), deferasirox (oral — most commonly used), deferiprone (oral). Monitor with serum ferritin and cardiac/hepatic MRI T2*. Allogeneic stem cell transplant is curative and is recommended for young patients with a matched sibling donor. Luspatercept (activin receptor ligand trap) reduces transfusion burden in beta-thal.
09 Acute Myeloid Leukemia (AML)
AML is a clonal hematopoietic neoplasm characterized by proliferation of immature myeloid precursors (blasts ≥20% in bone marrow or peripheral blood). It is the most common acute leukemia in adults, with a median age at diagnosis of ~68 years. Prognosis is determined by cytogenetics and molecular markers.
Presentation
Symptoms reflect marrow failure: anemia (fatigue, pallor), neutropenia (infections, fever), and thrombocytopenia (bleeding, petechiae). Gum hypertrophy and skin infiltration (leukemia cutis) are seen particularly in monocytic subtypes (M4/M5). DIC is characteristically associated with acute promyelocytic leukemia (APL / AML-M3).
Risk Stratification (ELN 2022)
| Risk | Cytogenetics / Molecular | Expected Outcomes |
|---|---|---|
| Favorable | t(8;21) RUNX1-RUNX1T1; inv(16)/t(16;16) CBFB-MYH11; NPM1 mutated without FLT3-ITD; biallelic CEBPA | CR 85–95%; 5-year OS 55–65%; chemo may be curative |
| Intermediate | Normal karyotype with NPM1+/FLT3-ITD+; t(9;11) KMT2A; cytogenetics not classified as favorable or adverse | CR 70–80%; 5-year OS 30–45%; consider transplant in CR1 |
| Adverse | Complex karyotype (≥3 abnormalities); monosomal karyotype; del(5q), -7/del(7q); TP53, RUNX1, ASXL1 mutations; FLT3-ITD high allelic ratio | CR 40–60%; 5-year OS 10–20%; transplant recommended |
Treatment
Fit patients (age <60–75, good PS): Induction with "7+3" — cytarabine continuous infusion × 7 days + daunorubicin or idarubicin × 3 days. If FLT3-mutated, add midostaurin. Consolidation: high-dose cytarabine (HiDAC) × 3–4 cycles for favorable risk; allogeneic stem cell transplant for intermediate/adverse risk in CR1.
Unfit patients: Venetoclax + azacitidine (now standard frontline for unfit patients per VIALE-A trial); low-dose cytarabine + venetoclax; targeted agents (IDH1/2 inhibitors: ivosidenib, enasidenib).
Acute promyelocytic leukemia with t(15;17) PML-RARA presents with life-threatening DIC. Treatment is all-trans retinoic acid (ATRA) + arsenic trioxide (ATO), which is curative in >90% of cases without conventional chemotherapy. Start ATRA immediately upon clinical suspicion — do not wait for molecular confirmation. Monitor for differentiation syndrome (fever, pulmonary infiltrates, weight gain) — treat with dexamethasone.
10 Acute Lymphoblastic Leukemia (ALL)
ALL is a neoplasm of lymphoid progenitors (B-cell or T-cell lineage) with ≥20% lymphoblasts in bone marrow. It is the most common malignancy in children (peak age 2–5 years) with cure rates of ~90% in pediatric patients. Adult ALL has a worse prognosis, with 5-year survival of 30–50%.
Classification & Risk Factors
| Feature | Details |
|---|---|
| B-cell ALL (~85%) | CD10 (CALLA)+, CD19+, CD22+; most common subtype |
| T-cell ALL (~15%) | CD3+, CD7+; often presents with mediastinal mass (thymic); more common in adolescent males |
| Philadelphia chromosome + (Ph+) | t(9;22) BCR-ABL; present in ~25% of adult B-ALL but only ~3% of pediatric ALL; historically very poor prognosis now improved with TKIs |
| Philadelphia-like (Ph-like) | Gene expression profile similar to Ph+ but lacks BCR-ABL; associated with kinase-activating mutations; poor prognosis |
| Favorable pediatric | Hyperdiploidy (>50 chromosomes), ETV6-RUNX1 t(12;21) |
| Adverse features | Hypodiploidy, KMT2A rearrangement, iAMP21, Ph+, age >35, WBC >30K (B-ALL) or >100K (T-ALL) |
Treatment Principles
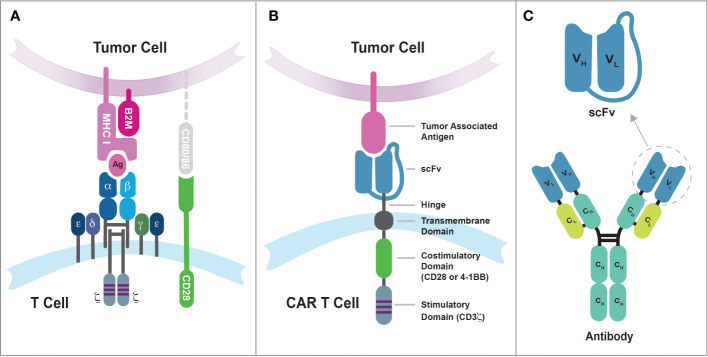
ALL treatment includes: induction (achieve CR: vincristine, dexamethasone/prednisone, PEG-asparaginase, +/- anthracycline), consolidation/intensification (high-dose methotrexate, cytarabine, cyclophosphamide), CNS prophylaxis (intrathecal methotrexate +/- cytarabine — ALL has high CNS relapse risk), and maintenance (oral methotrexate + 6-mercaptopurine for 2–3 years). Ph+ ALL adds a TKI (dasatinib preferred for CNS penetration). Blinatumomab (bispecific CD3/CD19 antibody) and inotuzumab (anti-CD22 ADC) are used in relapsed/refractory B-ALL.
CAR-T cell therapy: Tisagenlecleucel (Kymriah, anti-CD19) is FDA-approved for relapsed/refractory B-ALL in patients up to age 25. CR rates of 80–90% in heavily pretreated patients. Monitor for cytokine release syndrome (CRS) and neurotoxicity (ICANS); treat with tocilizumab (anti-IL-6R) and dexamethasone.
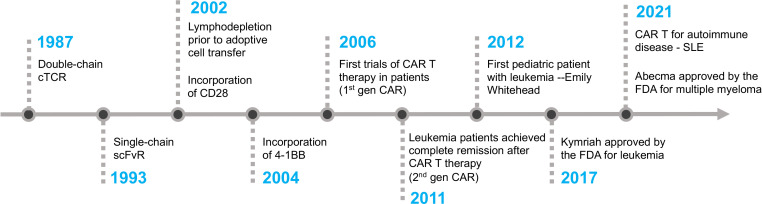
11 Chronic Myeloid Leukemia (CML)
CML is a myeloproliferative neoplasm defined by the Philadelphia chromosome t(9;22) producing the BCR-ABL1 fusion oncoprotein, a constitutively active tyrosine kinase. It accounts for ~15% of adult leukemias with a median age of diagnosis of ~64 years.
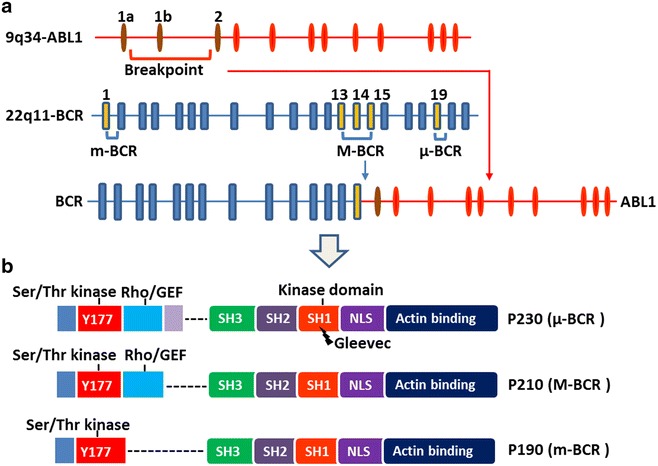
Clinical Phases
| Phase | Features | Blast % |
|---|---|---|
| Chronic phase (CP) | Leukocytosis with left shift (myelocytes, metamyelocytes); basophilia; splenomegaly; often asymptomatic | <10% in blood/marrow |
| Accelerated phase (AP) | Increasing WBC, basophilia ≥20%, additional cytogenetic abnormalities, treatment resistance | 10–19% |
| Blast crisis (BC) | Resembles acute leukemia (myeloid 60% or lymphoid 30%); poor prognosis | ≥20% |
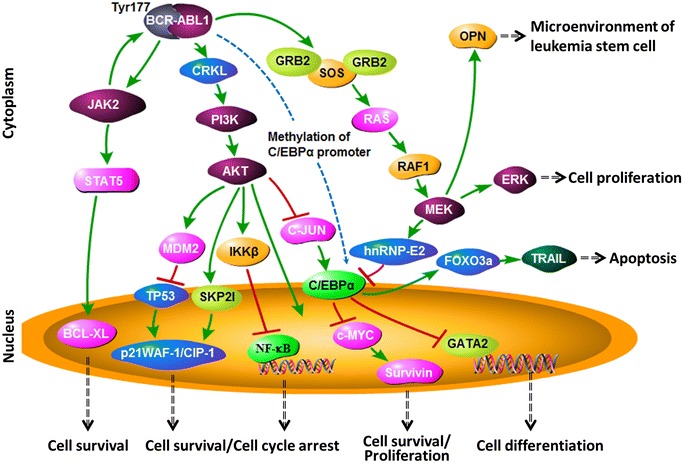
Tyrosine Kinase Inhibitor (TKI) Therapy
| TKI | Generation | Key Features |
|---|---|---|
| Imatinib (Gleevec) | 1st | First TKI; 400 mg daily; ~95% achieve complete hematologic response; well-tolerated; generic available |
| Dasatinib (Sprycel) | 2nd | More potent; active against most imatinib-resistant mutations (except T315I); risk of pleural effusion; good CNS penetration |
| Nilotinib (Tasigna) | 2nd | More selective BCR-ABL inhibitor; risk of cardiovascular events (PAD, QTc prolongation); take on empty stomach |
| Bosutinib (Bosulif) | 2nd | GI side effects common; option for intolerance to other TKIs |
| Ponatinib (Iclusig) | 3rd | Only TKI active against T315I gatekeeper mutation; risk of arterial occlusive events; reserve for T315I or multiply resistant |
| Asciminib (Scemblix) | STAMP inhibitor | Allosteric inhibitor (myristoyl pocket); active against T315I at higher dose; FDA-approved for CML-CP after ≥2 prior TKIs |
Monitoring & Treatment-Free Remission
Response milestones are monitored by BCR-ABL1 quantitative PCR (IS): major molecular response (MMR, BCR-ABL ≤0.1% IS) by 12 months, deep molecular response (MR4/MR4.5) for TFR eligibility. Treatment-free remission (TFR) can be attempted in patients with sustained deep molecular response (≥2 years of MR4 or deeper) on a TKI ≥3 years — approximately 50% maintain MMR off therapy. Monthly PCR monitoring is essential; TKI is restarted if molecular relapse occurs.
12 Chronic Lymphocytic Leukemia (CLL)
CLL is the most common leukemia in Western adults, characterized by clonal proliferation of mature-appearing CD5+ B lymphocytes. Median age at diagnosis is ~70 years. Many patients are asymptomatic and discovered incidentally on CBC showing lymphocytosis.
Diagnosis
Requires: persistent B-lymphocyte count ≥5 × 109/L for ≥3 months with characteristic immunophenotype: CD5+, CD19+, CD20 (dim), CD23+, surface Ig (dim). Peripheral smear shows mature lymphocytes with smudge cells. If lymph nodes are involved without peripheral lymphocytosis, it is classified as small lymphocytic lymphoma (SLL) — same disease, different presentation.
Staging
| Rai Stage | Features | Risk | Binet Stage |
|---|---|---|---|
| 0 | Lymphocytosis only | Low | A (<3 nodal areas) |
| I | + Lymphadenopathy | Intermediate | B (≥3 nodal areas) |
| II | + Splenomegaly/hepatomegaly | Intermediate | B |
| III | + Anemia (Hb <11 g/dL) | High | C (anemia and/or thrombocytopenia) |
| IV | + Thrombocytopenia (<100 × 109/L) | High | C |
Prognostic Markers
| Marker | Favorable | Adverse |
|---|---|---|
| IGHV mutation status | Mutated (indolent) | Unmutated (aggressive) |
| FISH cytogenetics | del(13q) isolated | del(17p), del(11q) |
| TP53 mutation | Absent | Present (chemo-refractory) |
| Beta-2 microglobulin | Low | Elevated |
Treatment
Indications to treat: symptomatic disease (B symptoms, fatigue), progressive cytopenias, massive/progressive lymphadenopathy or splenomegaly, autoimmune cytopenias refractory to steroids. Asymptomatic early-stage CLL is observed ("watch and wait").
| Agent Class | Examples | Notes |
|---|---|---|
| BTK inhibitors | Ibrutinib, acalabrutinib, zanubrutinib | First-line for most patients; continuous oral therapy; ibrutinib: AFib, bleeding, HTN; acalabrutinib/zanubrutinib: better tolerated; effective regardless of del(17p)/TP53 |
| BCL-2 inhibitor | Venetoclax (+/- obinutuzumab) | Fixed-duration therapy (12 months); requires TLS prophylaxis and dose ramp-up; first-line or relapsed; effective in del(17p) |
| Anti-CD20 antibodies | Rituximab, obinutuzumab, ofatumumab | Combined with chemo or venetoclax; obinutuzumab superior to rituximab in CLL |
| Chemoimmunotherapy | FCR (fludarabine, cyclophosphamide, rituximab) | Still considered for young, fit patients with IGHV-mutated CLL without del(17p); potentially curative in this subset |
13 Myelodysplastic Syndromes (MDS)
MDS are clonal hematopoietic stem cell disorders characterized by dysplastic morphology, ineffective hematopoiesis, and peripheral cytopenias with a risk of transformation to AML (approximately 30% over time). Median age at diagnosis is ~70 years. Most cases are de novo; therapy-related MDS occurs after prior chemotherapy (especially alkylating agents) or radiation.
IPSS-R (Revised International Prognostic Scoring System)
| Risk Group | Score | Median Survival | 25% AML Transformation |
|---|---|---|---|
| Very low | ≤1.5 | 8.8 years | Not reached |
| Low | >1.5–3.0 | 5.3 years | 10.8 years |
| Intermediate | >3.0–4.5 | 3.0 years | 3.2 years |
| High | >4.5–6.0 | 1.6 years | 1.4 years |
| Very high | >6.0 | 0.8 years | 0.7 years |
Management
| Risk | Approach |
|---|---|
| Lower-risk | Supportive care (transfusions, EPO/G-CSF); lenalidomide for del(5q) — 67% transfusion independence, cytogenetic response; luspatercept for ring sideroblasts with anemia |
| Higher-risk | Azacitidine or decitabine (hypomethylating agents) — improve survival and delay AML transformation; allogeneic stem cell transplant is the only curative option (for eligible patients) |
14 Hodgkin Lymphoma
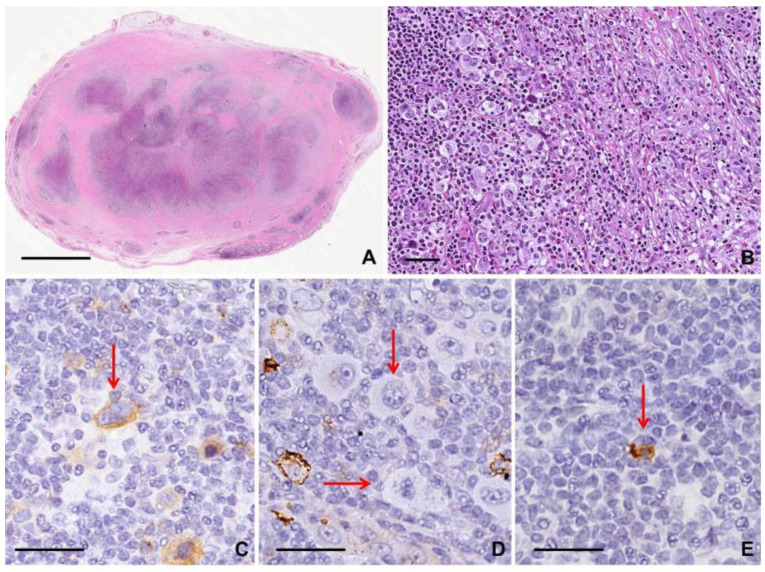
Hodgkin lymphoma (HL) accounts for ~10% of all lymphomas and has a bimodal age distribution (20–30 years and >55 years). It is defined by the presence of Reed-Sternberg cells (large, binucleated or multinucleated cells with prominent nucleoli — "owl-eye" appearance) in a reactive inflammatory background. RS cells are derived from germinal center B cells and typically express CD15+ and CD30+ but are CD20− (distinguishing from most NHL).
Ann Arbor Staging (Modified by Lugano)
| Stage | Description |
|---|---|
| I | Single lymph node region or single extralymphatic site (IE) |
| II | Two or more lymph node regions on the same side of the diaphragm |
| III | Lymph node regions on both sides of the diaphragm |
| IV | Diffuse or disseminated extralymphatic involvement (liver, bone marrow, lung) |
| Suffix A | No B symptoms |
| Suffix B | B symptoms present: fever >38°C, night sweats, weight loss >10% in 6 months |
Treatment
| Stage | Standard Treatment | Notes |
|---|---|---|
| Early favorable (I–II, no bulk) | ABVD × 2 cycles + ISRT (involved-site radiation) | Cure rate >95%; PET-adapted: if PET-negative after 2 cycles, may omit radiation |
| Early unfavorable (I–II, bulky or risk factors) | ABVD × 4–6 cycles +/- ISRT | Consider escalated BEACOPP or BV-AVD |
| Advanced (III–IV) | ABVD × 6 cycles or BV-AVD × 6 cycles | ECHELON-1 trial: brentuximab vedotin + AVD (BV-AVD) showed PFS benefit over ABVD in advanced HL; avoid bleomycin pulmonary toxicity |
| Relapsed/refractory | Salvage chemo (ICE/DHAP) → autologous SCT | Post-transplant brentuximab vedotin maintenance; checkpoint inhibitors (nivolumab, pembrolizumab) highly effective in R/R HL (ORR ~70%) |
15 Non-Hodgkin Lymphoma — Aggressive
Diffuse Large B-Cell Lymphoma (DLBCL)
DLBCL is the most common NHL subtype (~30–40% of all NHL). It presents as a rapidly growing mass (nodal or extranodal). Subtypes by gene expression profiling: germinal center B-cell (GCB, better prognosis) and activated B-cell (ABC, worse prognosis). IPI (International Prognostic Index) stratifies risk using age, stage, LDH, ECOG PS, and number of extranodal sites.
Treatment: R-CHOP × 6 cycles (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) is standard. Polatuzumab vedotin-R-CHP (Pola-R-CHP) showed superiority over R-CHOP in the POLARIX trial and is an emerging frontline option. For relapsed/refractory DLBCL, CAR-T cell therapy (axicabtagene ciloleucel, lisocabtagene maraleucel, tisagenlecleucel) is now standard in second line. Cure rate with R-CHOP: ~60–65%.
Burkitt Lymphoma
Highly aggressive B-cell NHL associated with t(8;14) MYC/IGH translocation. Three clinical variants: endemic (African, associated with EBV, jaw mass), sporadic (abdominal mass, most common in Western countries), and immunodeficiency-associated (HIV). "Starry sky" pattern on histology (macrophages phagocytosing apoptotic debris). Treatment: intensive chemo (R-CODOX-M/IVAC, R-hyper-CVAD, DA-R-EPOCH) with CNS prophylaxis. High tumor lysis risk — aggressive prophylaxis required.
Mantle Cell Lymphoma (MCL)
Characterized by t(11;14) CCND1/IGH translocation causing cyclin D1 overexpression. CD5+, CD20+, cyclin D1+. Median age ~65. Often presents at advanced stage. Indolent variant exists. Standard treatment: intensive chemo with rituximab (R-DHAP/cytarabine-based) followed by autologous SCT in fit patients; BTK inhibitors (ibrutinib, acalabrutinib, zanubrutinib) for relapsed disease; venetoclax-based combinations emerging.
Primary CNS Lymphoma
Almost always DLBCL. Associated with immunosuppression (HIV/AIDS). Presents with focal neurologic deficits, cognitive changes. MRI shows contrast-enhancing periventricular lesion(s). Diagnosis: stereotactic biopsy (avoid steroids before biopsy as they can lyse lymphoma cells). Treatment: high-dose methotrexate-based regimens (crosses blood-brain barrier); whole-brain radiation reserved for refractory cases due to neurocognitive toxicity.
Burkitt lymphoma, high-grade B-cell lymphoma, and any bulky aggressive NHL are high risk for tumor lysis syndrome. Start allopurinol or rasburicase, aggressive IV hydration, and monitor electrolytes (K+, phosphate, uric acid, calcium, creatinine) every 6–8 hours before and after initiating therapy.
16 Non-Hodgkin Lymphoma — Indolent
Follicular Lymphoma (FL)
Second most common NHL (~20%). Characterized by t(14;18) BCL2/IGH translocation causing BCL-2 overexpression (anti-apoptotic). Presents with painless generalized lymphadenopathy; bone marrow involvement is common. Grade 1–2 is indolent; grade 3A is treated as indolent or aggressive depending on context; grade 3B is treated as DLBCL. FLIPI (Follicular Lymphoma IPI) stratifies risk.
Management: Asymptomatic patients — watch and wait (observation does not worsen outcomes). When treatment is needed: rituximab monotherapy, bendamustine-rituximab (BR), or R-CHOP/R-CVP followed by rituximab maintenance × 2 years. Lenalidomide + rituximab (R2) is an effective chemo-free alternative. Histologic transformation to DLBCL occurs in ~3% per year and carries a worse prognosis.
Marginal Zone Lymphoma (MZL)
| Subtype | Association | Treatment |
|---|---|---|
| Extranodal (MALT) | Gastric MALT — H. pylori (70% respond to antibiotic eradication); ocular, thyroid, lung | H. pylori eradication for gastric MALT; radiation for localized; rituximab-based for advanced |
| Splenic MZL | HCV association | Splenectomy or rituximab; treat HCV if present |
| Nodal MZL | No specific association | Similar to follicular lymphoma management |
Waldenström Macroglobulinemia (WM)
Lymphoplasmacytic lymphoma with IgM monoclonal gammopathy. MYD88 L265P mutation found in >90%. Presents with hyperviscosity syndrome (blurred vision, headache, mucosal bleeding), peripheral neuropathy, cold agglutinins, cryoglobulinemia. Treatment (when symptomatic): BTK inhibitors (ibrutinib, zanubrutinib — ASPEN trial showed zanubrutinib better tolerated), bendamustine-rituximab. Avoid rituximab monotherapy upfront if IgM >4000 mg/dL (risk of IgM flare worsening hyperviscosity). Plasmapheresis for acute symptomatic hyperviscosity.
17 Multiple Myeloma
Multiple myeloma (MM) is a malignant plasma cell neoplasm characterized by monoclonal immunoglobulin production, bone destruction, and end-organ damage. Median age at diagnosis is ~69 years. It accounts for ~10% of hematologic malignancies.
CRAB Criteria (End-Organ Damage)
| Criterion | Definition | Mechanism |
|---|---|---|
| Calcium elevation | Serum calcium >11 mg/dL or >1 mg/dL above ULN | Osteoclast activation by myeloma cells |
| Renal insufficiency | Creatinine >2 mg/dL or CrCl <40 mL/min | Light chain cast nephropathy (myeloma kidney), hypercalcemia, amyloidosis |
| Anemia | Hemoglobin <10 g/dL or >2 g/dL below LLN | Marrow infiltration by plasma cells, EPO suppression |
| Bone disease | Lytic lesions on skeletal survey, CT, or PET/CT | RANKL/OPG imbalance; osteoclast activation with osteoblast suppression |
SLiM-CRAB Criteria (Myeloma-Defining Events)
In addition to CRAB, the following are now considered myeloma-defining events warranting treatment even without CRAB: Sixty percent or more clonal plasma cells in marrow; Light chain ratio ≥100 (involved/uninvolved); MRI with >1 focal lesion (≥5 mm).
Diagnostic Workup
SPEP/UPEP with immunofixation, serum free light chains, CBC, calcium, creatinine, LDH, beta-2 microglobulin, albumin, bone marrow biopsy with cytogenetics/FISH, whole-body low-dose CT or PET/CT (skeletal survey is now less preferred).
Staging (R-ISS)
| Stage | Criteria | Median OS |
|---|---|---|
| I | Beta-2M <3.5 mg/L + albumin ≥3.5 g/dL + standard-risk cytogenetics + normal LDH | Not reached (~82% 5-yr OS) |
| II | Not stage I or III | ~83 months |
| III | Beta-2M ≥5.5 mg/L + high-risk cytogenetics [t(4;14), t(14;16), del(17p)] and/or elevated LDH | ~43 months |
Treatment
| Setting | Regimen | Notes |
|---|---|---|
| Transplant-eligible (age <65–70, fit) | Induction: VRd (bortezomib, lenalidomide, dexamethasone) × 3–4 cycles → autologous SCT → lenalidomide maintenance | DETERMINATION and IFM 2009 trials established this approach; isatuximab- or daratumumab-VRd emerging as new standard (GRIFFIN, PERSEUS trials) |
| Transplant-ineligible | VRd or DRd (daratumumab, lenalidomide, dexamethasone) until progression | MAIA trial: DRd superior to Rd in transplant-ineligible |
| Relapsed/refractory | Carfilzomib, pomalidomide, daratumumab, isatuximab, elotuzumab, selinexor, belantamab mafodotin, bispecific antibodies (teclistamab), CAR-T (idecabtagene vicleucel, ciltacabtagene autoleucel) | BCMA-targeting agents represent major advance; CAR-T in 4th+ line showing deep responses |
18 MGUS & Smoldering Myeloma
Monoclonal Gammopathy of Undetermined Significance (MGUS)
| Feature | Criteria |
|---|---|
| Definition | M-protein <3 g/dL, bone marrow plasma cells <10%, no end-organ damage (no CRAB or SLiM criteria) |
| Prevalence | ~3–4% of population >50 years; increases with age |
| Progression risk | ~1% per year to myeloma or related malignancy (lifelong risk) |
| Risk factors for progression | Non-IgG isotype, M-protein >1.5 g/dL, abnormal free light chain ratio |
| Monitoring | SPEP, CBC, calcium, creatinine every 6–12 months; no treatment indicated |
Smoldering Multiple Myeloma (SMM)
| Feature | Criteria |
|---|---|
| Definition | M-protein ≥3 g/dL and/or bone marrow plasma cells 10–59%, no myeloma-defining events |
| Progression risk | ~10% per year for first 5 years, then decreasing; overall ~50% at 5 years for high-risk SMM |
| 20/2/20 high-risk model | ≥2 of: M-protein >2 g/dL, FLC ratio >20, BMPC >20% → high risk (~50% progress within 2 years) |
| Management | Standard: close observation with labs every 2–3 months. Clinical trials of early intervention for high-risk SMM (lenalidomide-dexamethasone showed PFS benefit in QUIREDEX and E3A06 trials) |
19 AL Amyloidosis
AL (immunoglobulin light chain) amyloidosis results from deposition of misfolded monoclonal light chain fragments (usually lambda) as insoluble amyloid fibrils in tissues. It is a systemic disease that can affect virtually any organ. It is related to plasma cell neoplasms but requires its own distinct management approach.
Organ Involvement
| Organ | Manifestations | Key Findings |
|---|---|---|
| Heart (~75%) | Restrictive cardiomyopathy, diastolic heart failure, arrhythmias | Thickened LV wall on echo but low voltage on ECG (pathognomonic mismatch); elevated NT-proBNP and troponin |
| Kidney (~65%) | Nephrotic syndrome (massive proteinuria, edema) | Albuminuria (not Bence Jones proteinuria) |
| Liver | Hepatomegaly, elevated alkaline phosphatase | Cholestatic pattern without biliary obstruction |
| GI tract | Macroglossia (almost pathognomonic), GI bleeding, malabsorption | Macroglossia + periorbital purpura = classic presentation |
| Peripheral nervous system | Peripheral neuropathy, autonomic neuropathy (orthostatic hypotension), carpal tunnel | Bilateral carpal tunnel preceding systemic diagnosis is common |
| Soft tissue | Periorbital purpura ("raccoon eyes"), factor X deficiency | Purpura after minor trauma or Valsalva |
Diagnosis & Treatment
Diagnosis: Tissue biopsy showing apple-green birefringence under polarized light with Congo red staining. Fat pad aspirate is least invasive (~80% sensitivity). Mass spectrometry confirms AL subtype. Must also demonstrate a clonal plasma cell disorder (SPEP/immunofixation, serum free light chains, bone marrow biopsy).
Treatment: Directed at the underlying plasma cell clone. Daratumumab-VCd (daratumumab, bortezomib, cyclophosphamide, dexamethasone) is the new standard based on the ANDROMEDA trial. Autologous SCT for selected patients (no severe cardiac involvement). Organ response is monitored by NT-proBNP (cardiac) and proteinuria (renal).
Cardiac involvement is the primary determinant of survival in AL amyloidosis. Digoxin is contraindicated as amyloid fibrils bind digoxin, increasing toxicity risk. Similarly, calcium channel blockers can worsen heart failure. Suspect cardiac AL in any patient with unexplained heart failure + thick ventricles + low-voltage ECG.
20 Hemostasis Overview
Hemostasis is the physiologic process that stops bleeding while maintaining blood fluidity. It involves three phases: primary hemostasis (platelet plug formation), secondary hemostasis (coagulation cascade forming fibrin clot), and fibrinolysis (clot breakdown by plasmin).
Primary Hemostasis
Vascular injury exposes subendothelial collagen and von Willebrand factor (vWF). Platelets adhere via the GPIb-vWF interaction, become activated (releasing ADP, thromboxane A2, serotonin from dense and alpha granules), and aggregate via GPIIb/IIIa receptor binding fibrinogen. Defects in primary hemostasis cause mucocutaneous bleeding: petechiae, epistaxis, gingival bleeding, menorrhagia. Platelet count and bleeding time (or PFA-100) assess primary hemostasis.
Secondary Hemostasis (Coagulation Cascade)
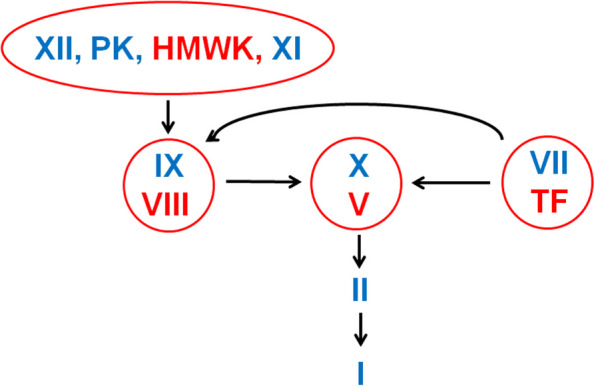
PT/INR vs aPTT — What Each Measures
| Test | Pathway | Factors Assessed | Prolonged By |
|---|---|---|---|
| PT / INR | Extrinsic + common | VII, X, V, II, fibrinogen | Warfarin, vitamin K deficiency, liver disease, factor VII deficiency |
| aPTT | Intrinsic + common | XII, XI, IX, VIII, X, V, II, fibrinogen | Heparin (UFH), hemophilia A (VIII) / B (IX), lupus anticoagulant, factor XII deficiency |
| Both prolonged | Common pathway or systemic | X, V, II, fibrinogen | DIC, liver disease, supratherapeutic anticoagulation, severe vitamin K deficiency |
Primary vs Secondary Hemostasis Defects
| Feature | Primary (Platelet/vWF) | Secondary (Coagulation Factor) |
|---|---|---|
| Bleeding pattern | Mucocutaneous: petechiae, purpura, epistaxis, gingival, menorrhagia | Deep tissue: hemarthrosis, muscle hematomas, retroperitoneal |
| Onset after injury | Immediate | Delayed (hours) |
| Response to pressure | Effective | Temporary, rebleeds |
| Lab findings | Low platelets or prolonged PFA-100/bleeding time | Prolonged PT and/or aPTT |
21 Thrombocytopenia
Thrombocytopenia is defined as a platelet count <150 × 109/L. The approach is to categorize by mechanism: decreased production, increased destruction, or sequestration. Always rule out pseudothrombocytopenia (EDTA-dependent platelet clumping) by examining the smear or repeating in a citrate tube.
Key Causes of Thrombocytopenia
| Condition | Mechanism | Key Features | Management |
|---|---|---|---|
| ITP | Autoimmune platelet destruction (anti-GPIIb/IIIa antibodies) + impaired thrombopoiesis | Diagnosis of exclusion; isolated thrombocytopenia; normal smear except low platelets; no splenomegaly | Steroids (dexamethasone or prednisone) first-line; IVIG for acute bleeding; rituximab, TPO-agonists (romiplostim, eltrombopag) for refractory; splenectomy |
| TTP | ADAMTS13 deficiency (<10%) → large vWF multimers → platelet microthrombi | Pentad: thrombocytopenia, MAHA (schistocytes), fever, renal dysfunction, neurologic changes. Classic pentad in <5%; thrombocytopenia + MAHA is sufficient to act | Emergent plasma exchange (PLEX); steroids; caplacizumab (anti-vWF); do NOT transfuse platelets (may worsen thrombosis) |
| HIT | Anti-PF4/heparin antibodies → platelet activation and consumption | Platelet drop >50% or to <150K, 5–10 days after heparin exposure; paradoxical thrombosis | See Section 27 |
| DIC | Systemic activation of coagulation → consumptive coagulopathy | Schistocytes, elevated PT/aPTT, low fibrinogen, elevated D-dimer; often in sepsis, malignancy, obstetric emergencies | Treat underlying cause; supportive: platelets, FFP, cryoprecipitate for active bleeding; heparin only in chronic DIC with thrombosis |
| Drug-induced | Various (immune, myelosuppressive) | Common culprits: chemotherapy, valproic acid, linezolid, vancomycin, quinine, sulfonamides | Discontinue offending agent |
| Splenic sequestration | Up to 90% of platelets sequestered in enlarged spleen | Liver cirrhosis with portal hypertension; typically mild (50–100K) | Treat underlying liver disease; rarely needs intervention |
If a patient presents with unexplained thrombocytopenia and microangiopathic hemolytic anemia (schistocytes on smear), presume TTP and initiate plasma exchange immediately. Mortality of untreated TTP exceeds 90%. Send ADAMTS13 activity level but do not wait for results to begin treatment. Do NOT transfuse platelets.
22 Inherited Bleeding Disorders
Hemophilia A & B
| Feature | Hemophilia A | Hemophilia B |
|---|---|---|
| Deficient factor | Factor VIII | Factor IX |
| Inheritance | X-linked recessive | X-linked recessive |
| Frequency | 1 in 5,000 males | 1 in 30,000 males |
| Lab findings | Prolonged aPTT, normal PT, low factor VIII level | Prolonged aPTT, normal PT, low factor IX level |
| Severity classification | Severe: <1% factor activity (spontaneous bleeds); Moderate: 1–5% (bleeding with minor trauma); Mild: 5–40% (bleeding with surgery/major trauma) | |
| Treatment | Factor VIII concentrate (recombinant preferred); DDAVP for mild hemophilia A (releases vWF/VIII from endothelium); emicizumab (bispecific antibody mimicking factor VIII) for prophylaxis | Factor IX concentrate; extended half-life products available |
| Inhibitors | Alloantibodies that neutralize infused factor; occurs in ~25–30% of severe hemophilia A; treat acute bleeds with bypassing agents (rFVIIa, FEIBA); immune tolerance induction (ITI) | |
Von Willebrand Disease (vWD)
vWD is the most common inherited bleeding disorder (~1% prevalence). vWF mediates platelet adhesion (GPIb binding) and stabilizes factor VIII in circulation.
| Type | Defect | Frequency | Features | Treatment |
|---|---|---|---|---|
| Type 1 (~70–80%) | Partial quantitative deficiency | Most common | Mild mucocutaneous bleeding; low vWF antigen and activity | DDAVP (desmopressin) — first-line; releases stored vWF; vWF concentrate if DDAVP insufficient |
| Type 2 (~15–20%) | Qualitative defect (2A, 2B, 2M, 2N subtypes) | Variable | Disproportionately low vWF activity relative to antigen; 2B — gain-of-function with thrombocytopenia; 2N — decreased VIII binding (mimics hemophilia A) | vWF concentrate preferred (DDAVP may worsen thrombocytopenia in type 2B) |
| Type 3 (<5%) | Complete absence of vWF | Rare; autosomal recessive | Severe bleeding (mucocutaneous + deep tissue due to very low VIII) | vWF/VIII concentrate |
23 Acquired Coagulopathies
Disseminated Intravascular Coagulation (DIC)
DIC is a consumptive coagulopathy triggered by systemic activation of the coagulation cascade. It results in simultaneous widespread microvascular thrombosis and hemorrhagic diathesis from consumption of platelets and clotting factors.
| Feature | Details |
|---|---|
| Triggers | Sepsis (most common), trauma/burns, obstetric emergencies (placental abruption, amniotic fluid embolism), malignancy (APL, mucin-secreting adenocarcinomas), snake envenomation |
| Lab findings | Thrombocytopenia, prolonged PT/aPTT, low fibrinogen, elevated D-dimer, schistocytes on smear |
| Acute (overt) DIC | Bleeding predominates; consumptive; treat underlying cause + replace products (platelets if <10K or active bleeding, FFP for prolonged PT, cryoprecipitate if fibrinogen <100 mg/dL) |
| Chronic (compensated) DIC | Thrombosis predominates; liver compensates by producing factors; consider heparin; seen in malignancy (Trousseau syndrome) |
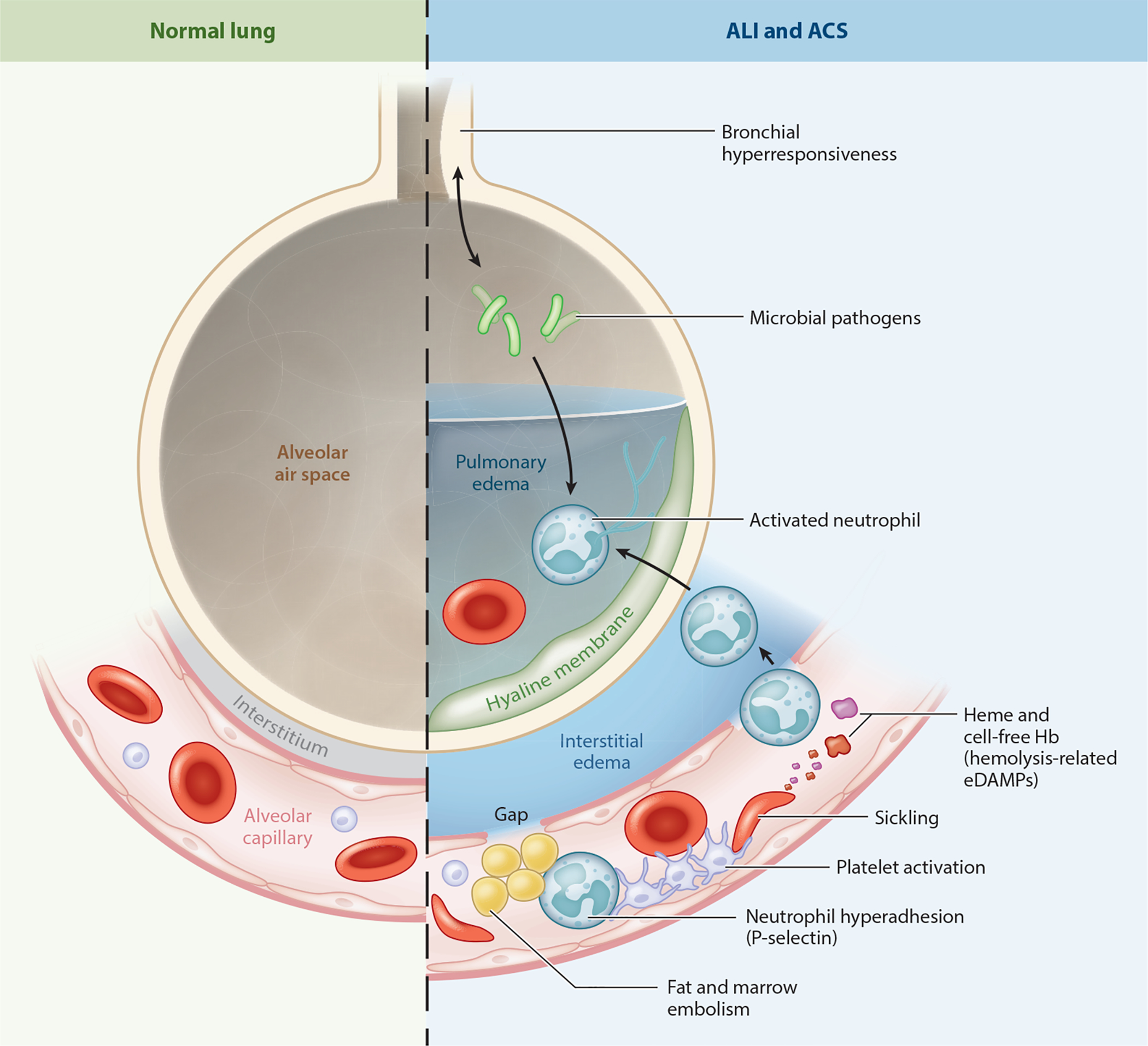
The cornerstone of DIC treatment is treating the underlying cause. Supportive blood product replacement is indicated for active bleeding or planned procedures: platelets (goal >50K if bleeding), FFP (for coagulation factors if PT >1.5× normal), cryoprecipitate (if fibrinogen <100–150 mg/dL). Do not withhold products out of concern for "fueling the fire" — this is a myth.
Liver Disease Coagulopathy
The liver synthesizes virtually all clotting factors (except vWF and factor VIII, which may actually be elevated). Liver disease causes a "rebalanced" hemostasis with reduced procoagulant and anticoagulant factors. PT/INR is prolonged first (factor VII has the shortest half-life). INR in liver disease does NOT predict bleeding risk as it does in warfarin therapy. Manage with FFP or prothrombin complex concentrate (PCC) only for active bleeding or pre-procedure; vitamin K if cholestasis suspected.
Vitamin K Deficiency
Vitamin K is required for gamma-carboxylation of factors II, VII, IX, X, and proteins C and S. Deficiency occurs in malnutrition, prolonged antibiotic use (disrupts gut flora), malabsorption, and neonates. Lab: prolonged PT initially (factor VII first to decline due to shortest half-life ~6 hours), then aPTT. Treatment: vitamin K 10 mg IV/PO (IV if urgent; onset 6–8 hours IV, 24 hours PO); FFP/PCC for life-threatening bleeding.
Acquired Hemophilia
Rare autoimmune condition with development of autoantibodies against factor VIII. Presents in elderly patients or postpartum with severe bleeding and isolated prolonged aPTT that does NOT correct with mixing study. Treatment: bypassing agents (rFVIIa, FEIBA) for acute bleeds; immunosuppression (steroids + cyclophosphamide or rituximab) to eradicate the inhibitor.
24 Anticoagulation Management
Anticoagulant Agents
| Agent | Mechanism | Monitoring | Reversal | Key Pearls |
|---|---|---|---|---|
| Unfractionated heparin (UFH) | Activates antithrombin III → inhibits thrombin (IIa) and Xa | aPTT (target 1.5–2.5× normal) or anti-Xa level | Protamine sulfate (1 mg per 100 units heparin; full reversal) | Short half-life (60–90 min); preferred when rapid on/off needed (peri-procedural, renal failure); HIT risk |
| LMWH (enoxaparin) | Primarily anti-Xa (via antithrombin) | Generally no monitoring needed; anti-Xa level in obesity, renal impairment, pregnancy | Protamine (60–80% reversal) | Renally cleared — dose-adjust or avoid if CrCl <30; predictable pharmacokinetics; lower HIT risk than UFH |
| Warfarin | Vitamin K antagonist (inhibits II, VII, IX, X, protein C/S) | INR (target 2–3 for most indications; 2.5–3.5 for mechanical mitral valve) | Vitamin K (PO/IV), FFP, 4-factor PCC (Kcentra) for urgent reversal | Narrow therapeutic index; many drug/food interactions; CYP2C9/VKORC1 pharmacogenomics; bridge with heparin initially (protein C drops first → transient hypercoagulability) |
| Rivaroxaban | Direct factor Xa inhibitor | No routine monitoring; anti-Xa calibrated if needed | Andexanet alfa (Andexxa) | Take with food (15 mg and 20 mg doses); once or twice daily depending on indication; avoid if CrCl <15 |
| Apixaban | Direct factor Xa inhibitor | No routine monitoring | Andexanet alfa | Safest DOAC in renal impairment (minimal renal clearance); BID dosing; dose reduce if ≥2 of: age ≥80, weight ≤60 kg, Cr ≥1.5 |
| Dabigatran | Direct thrombin (IIa) inhibitor | No routine monitoring; dTT or ecarin time if needed | Idarucizumab (Praxbind) — specific reversal agent | Most renally cleared DOAC; contraindicated if CrCl <30; GI side effects; can be removed by dialysis |
| Edoxaban | Direct factor Xa inhibitor | No routine monitoring | Andexanet alfa | Once daily; avoid if CrCl >95 (paradoxically less effective); dose reduce if CrCl 15–50 |
25 Venous Thromboembolism
Venous thromboembolism (VTE) encompasses deep vein thrombosis (DVT) and pulmonary embolism (PE). It is the third most common cardiovascular disease after MI and stroke, with annual incidence of ~1–2 per 1,000 adults.
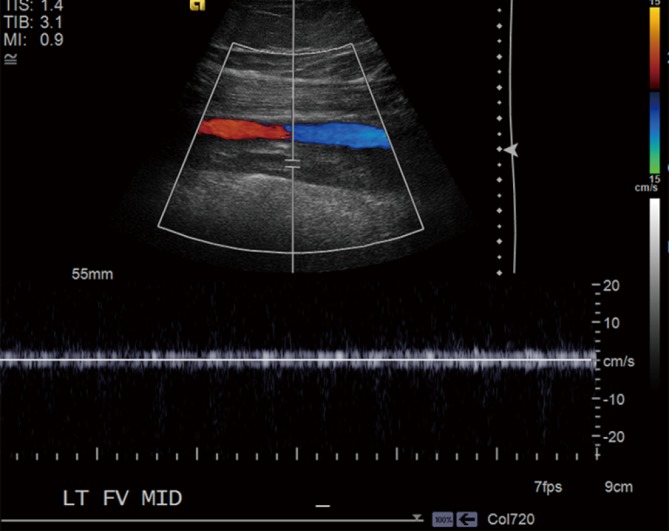
Wells Score for DVT
| Criterion | Points |
|---|---|
| Active cancer (treatment within 6 months or palliative) | +1 |
| Paralysis, paresis, or recent cast of lower extremity | +1 |
| Bedridden >3 days or major surgery within 12 weeks | +1 |
| Localized tenderness along deep venous system | +1 |
| Entire leg swollen | +1 |
| Calf swelling >3 cm (compared to asymptomatic leg) | +1 |
| Pitting edema (greater in symptomatic leg) | +1 |
| Collateral superficial veins (non-varicose) | +1 |
| Previously documented DVT | +1 |
| Alternative diagnosis as likely or more likely than DVT | −2 |
Score ≤1: DVT unlikely → check D-dimer (if negative, DVT excluded). Score ≥2: DVT likely → compression ultrasound. Age-adjusted D-dimer (age × 10 ng/mL for patients >50) improves specificity.
Treatment Duration for VTE
| Clinical Scenario | Duration | Notes |
|---|---|---|
| Provoked VTE (surgery, immobilization, estrogen) | 3 months | Low recurrence risk after provoking factor resolved |
| Unprovoked first VTE | ≥3 months, then reassess | Consider indefinite anticoagulation if low bleeding risk; D-dimer after stopping can guide decision |
| Recurrent unprovoked VTE | Indefinite | High recurrence risk off anticoagulation |
| Cancer-associated VTE | Indefinite (while cancer active) | LMWH or DOACs (edoxaban, rivaroxaban per SELECT-D, Hokusai VTE-Cancer); caution with DOACs in GI/GU cancers (higher mucosal bleeding) |
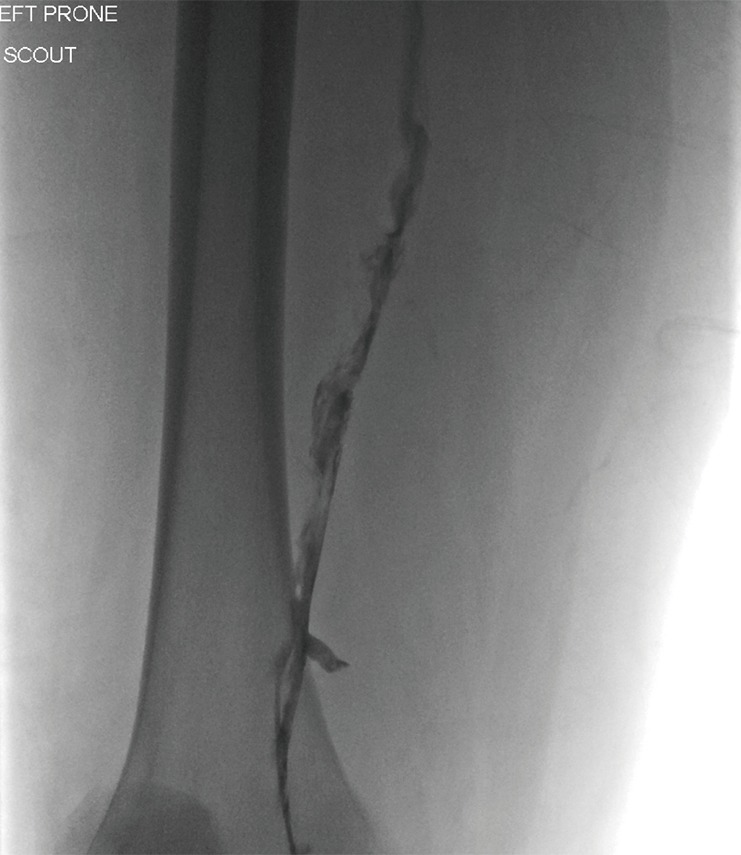
Massive PE
Massive PE (with hemodynamic instability: SBP <90 mmHg) requires systemic thrombolysis (alteplase 100 mg IV over 2 hours) or catheter-directed therapy/surgical embolectomy if thrombolysis is contraindicated. Submassive PE (RV strain without hypotension) may benefit from escalated therapy on a case-by-case basis.
26 Hypercoagulable States
Inherited Thrombophilias
| Condition | Defect | Prevalence | VTE Risk |
|---|---|---|---|
| Factor V Leiden | Resistance to activated protein C (factor V R506Q mutation) | 5% of Caucasians (heterozygous) | 3–8× (heterozygous); 50–80× (homozygous) |
| Prothrombin G20210A | Elevated prothrombin levels | 2–3% of Caucasians | 2–5× |
| Protein C deficiency | Reduced natural anticoagulant | 0.2–0.5% | 7–10×; warfarin-induced skin necrosis risk |
| Protein S deficiency | Reduced protein C cofactor | 0.1–1% | 5–10× |
| Antithrombin deficiency | Reduced heparin cofactor | 0.02–0.2% | 10–50× (highest risk inherited thrombophilia); may cause heparin resistance |
Antiphospholipid Syndrome (APS)
APS is an acquired autoimmune thrombophilia defined by thrombosis or pregnancy morbidity in the presence of persistent antiphospholipid antibodies (positive on ≥2 occasions, ≥12 weeks apart): lupus anticoagulant (paradoxically prolongs aPTT in vitro but causes thrombosis in vivo), anticardiolipin antibodies (IgG/IgM), and anti-beta2 glycoprotein I antibodies. Treatment: warfarin (INR 2–3) for thrombotic APS. DOACs are not recommended for APS (TRAPS trial showed increased thrombosis with rivaroxaban vs warfarin, especially in triple-positive APS).
When to Test for Thrombophilia
Testing is most useful in: unprovoked VTE at young age (<50), recurrent VTE, VTE at unusual sites (cerebral, splanchnic, hepatic), family history of VTE, and recurrent pregnancy loss. Do NOT test during acute thrombosis or on anticoagulation (results are unreliable). Testing rarely changes management of the acute event but may influence anticoagulation duration.
27 Heparin-Induced Thrombocytopenia
HIT is a prothrombotic immune-mediated drug reaction caused by IgG antibodies against the platelet factor 4 (PF4)–heparin complex. These antibodies activate platelets, generating a massive prothrombotic state. HIT occurs in ~0.5–5% of patients on UFH (less common with LMWH). Despite thrombocytopenia, the major risk is thrombosis (30–50% if untreated), not bleeding.
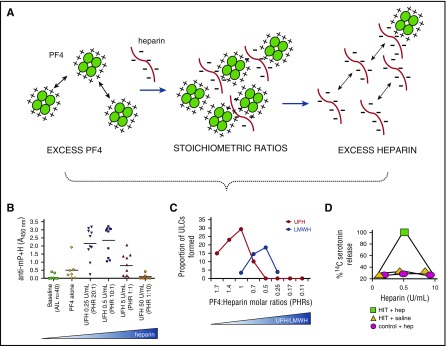
4T Score for Pretest Probability
| Criterion | 2 Points | 1 Point | 0 Points |
|---|---|---|---|
| Thrombocytopenia | Fall >50% and nadir ≥20K | Fall 30–50% or nadir 10–19K | Fall <30% or nadir <10K |
| Timing of platelet fall | Days 5–10, or ≤1 day if prior heparin within 30 days | Consistent with days 5–10 but unclear; onset >10 days | ≤4 days without recent exposure |
| Thrombosis or other sequelae | New thrombosis, skin necrosis, or acute systemic reaction | Progressive or recurrent thrombosis; erythematous skin lesions | None |
| Other cause for Thrombocytopenia | None apparent | Possible | Definite |
Score 0–3: Low probability (<5% HIT) — HIT unlikely. Score 4–5: Intermediate. Score 6–8: High probability. If intermediate or high probability: send PF4/heparin ELISA (high sensitivity, moderate specificity) and confirmatory serotonin release assay (SRA, gold standard). Stop all heparin immediately and start alternative anticoagulation.
Alternative Anticoagulants for HIT
| Agent | Mechanism | Notes |
|---|---|---|
| Argatroban | Direct thrombin inhibitor (IV) | Hepatically metabolized; preferred in renal failure; prolongs INR (complicates warfarin transition) |
| Bivalirudin | Direct thrombin inhibitor (IV) | Short half-life; used in cardiac surgery/PCI setting |
| Fondaparinux | Synthetic indirect Xa inhibitor (SC) | Does not cross-react with HIT antibodies; commonly used off-label; no monitoring needed |
| DOACs | Direct Xa or thrombin inhibitors (PO) | Emerging evidence for use in acute HIT once platelet count recovering; not yet standard initial therapy |
1) Stop ALL heparin (including flushes, heparin-coated catheters). 2) Start a non-heparin anticoagulant immediately (even without active thrombosis — the thrombotic risk is very high). 3) Do NOT transfuse platelets (may worsen thrombosis). 4) Do NOT start warfarin until platelets have recovered to ≥150K (risk of venous limb gangrene from protein C depletion).
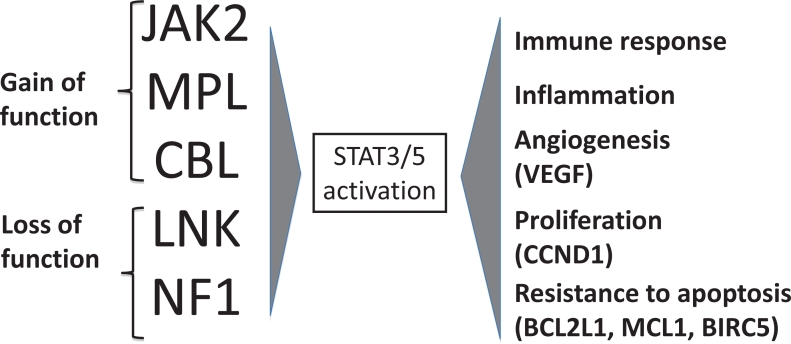
28 Polycythemia Vera
Polycythemia vera (PV) is a clonal myeloproliferative neoplasm characterized by erythrocytosis (elevated red cell mass) driven by the JAK2 V617F mutation (present in >95% of PV cases; JAK2 exon 12 mutations in remaining). The major risks are thrombosis (stroke, MI, DVT, splanchnic vein thrombosis) and transformation to myelofibrosis or AML.
Diagnosis (WHO 2022 Criteria)
| Major Criteria | Minor Criterion |
|---|---|
| 1. Hb >16.5 g/dL (men) or >16 g/dL (women), or Hct >49% (men) / >48% (women), or increased red cell mass | Subnormal serum EPO level |
| 2. Bone marrow biopsy: hypercellularity, trilineage proliferation (panmyelosis), pleomorphic megakaryocytes | — |
| 3. JAK2 V617F or JAK2 exon 12 mutation | — |
Diagnosis: all 3 major criteria, OR first 2 major + the minor criterion.
Management
| All patients | High-risk patients (age ≥60 OR prior thrombosis) |
|---|---|
| Phlebotomy to target Hct <45% (CYTO-PV trial); low-dose aspirin 81 mg daily (ECLAP trial) | Add hydroxyurea (first-line cytoreductive); alternatives: pegylated interferon-alpha (preferred in younger patients, pregnancy-safe), busulfan, ruxolitinib (JAK inhibitor — for hydroxyurea-intolerant/resistant) |
29 Essential Thrombocythemia
Essential thrombocythemia (ET) is an MPN characterized by sustained thrombocytosis (≥450 × 109/L) with megakaryocyte proliferation. Driver mutations: JAK2 V617F (~60%), CALR (~25%), MPL (~5%), and triple-negative (~10%). Risks include thrombosis and, less commonly, hemorrhage (especially with extreme thrombocytosis >1,000K due to acquired von Willebrand syndrome).
Risk Stratification (IPSET-Thrombosis)
| Risk | Criteria | Management |
|---|---|---|
| Very low | Age <60, no prior thrombosis, JAK2 wild-type | Observation only |
| Low | Age <60, no prior thrombosis, JAK2 mutated | Low-dose aspirin |
| Intermediate | Age ≥60, no prior thrombosis, JAK2 wild-type | Low-dose aspirin or observation |
| High | Age ≥60 with JAK2 mutation, OR prior thrombosis at any age | Cytoreduction (hydroxyurea first-line) + low-dose aspirin |
30 Primary Myelofibrosis
Primary myelofibrosis (PMF) is an MPN characterized by progressive bone marrow fibrosis, extramedullary hematopoiesis (especially in the spleen, causing massive splenomegaly), and a leukoerythroblastic blood picture (teardrop cells, nucleated RBCs, immature granulocytes). Driver mutations: JAK2 V617F (~60%), CALR (~25%), MPL (~5%). It carries the worst prognosis of the classic BCR-ABL-negative MPNs.
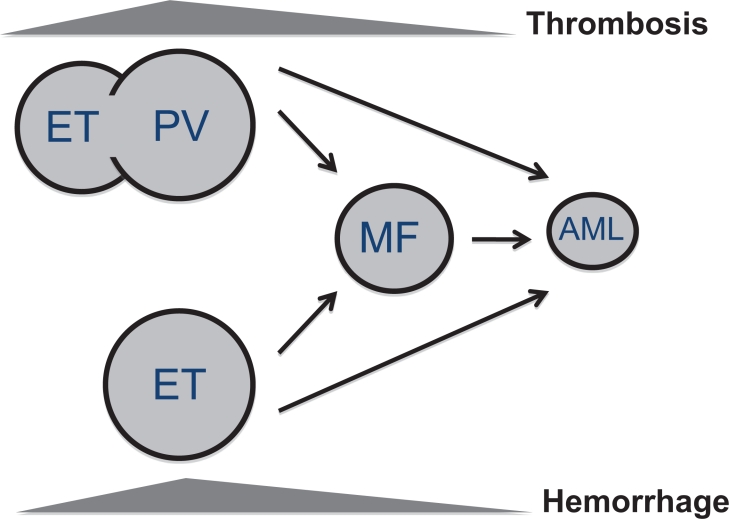
Clinical Features
Constitutional symptoms (fatigue, night sweats, weight loss, bone pain), massive splenomegaly (can cause early satiety, portal hypertension), cytopenias (especially anemia), leukoerythroblastic smear with teardrop cells (dacrocytes). "Dry tap" on bone marrow aspiration due to fibrosis; biopsy shows reticulin/collagen fibrosis.
Prognosis & Management
| Approach | Details |
|---|---|
| Risk stratification | DIPSS/DIPSS-Plus: age >65, constitutional symptoms, Hb <10, WBC >25K, circulating blasts ≥1%, unfavorable karyotype, transfusion dependence, platelets <100K |
| Low/intermediate-1 risk | Observation; manage anemia (EPO, danazol, lenalidomide); ruxolitinib for symptomatic splenomegaly or constitutional symptoms (COMFORT trials) |
| Intermediate-2/high risk | Allogeneic stem cell transplant (only curative therapy); ruxolitinib as bridge to transplant or for non-transplant candidates; fedratinib (JAK2 inhibitor) for ruxolitinib-refractory |
| Splenomegaly management | Ruxolitinib (reduces spleen size ~35% in COMFORT-I); splenectomy or splenic radiation for refractory cases |
31 Cancer Staging & Performance Status
TNM Staging System
| Component | Description | Categories |
|---|---|---|
| T (Tumor) | Size and extent of primary tumor | T0 (no primary tumor), Tis (carcinoma in situ), T1–T4 (increasing size/invasion) |
| N (Nodes) | Regional lymph node involvement | N0 (no nodes), N1–N3 (increasing nodal involvement) |
| M (Metastasis) | Distant metastasis | M0 (no metastasis), M1 (distant metastasis present) |
ECOG Performance Status
| Grade | Description |
|---|---|
| 0 | Fully active; no restrictions |
| 1 | Restricted in strenuous activity; ambulatory and able to carry out light work |
| 2 | Ambulatory and capable of self-care; unable to work; up and about >50% of waking hours |
| 3 | Capable of only limited self-care; confined to bed or chair >50% of waking hours |
| 4 | Completely disabled; totally confined to bed or chair; cannot carry on any self-care |
| 5 | Dead |
Common Tumor Markers
| Marker | Associated Cancer | Clinical Use |
|---|---|---|
| PSA | Prostate | Screening (controversial), monitoring treatment response |
| CA-125 | Ovarian | Monitoring, not screening (elevated in many benign conditions) |
| CA 19-9 | Pancreatic, biliary | Monitoring; may be elevated in pancreatitis, cholangitis |
| CEA | Colorectal | Monitoring for recurrence post-resection; not for screening |
| AFP | Hepatocellular carcinoma, testicular (nonseminoma) | Screening (HCC in cirrhosis), staging, monitoring |
| Beta-hCG | Testicular (choriocarcinoma, nonseminoma), gestational trophoblastic disease | Diagnosis, staging, monitoring |
| LDH | Lymphoma, melanoma, testicular | Prognostic, monitoring (nonspecific) |
32 Chemotherapy Principles
Cell Cycle & Drug Classes
| Class | Examples | Mechanism | Key Toxicities |
|---|---|---|---|
| Alkylating agents | Cyclophosphamide, ifosfamide, melphalan, busulfan, temozolomide | Cross-link DNA; cell cycle-independent | Myelosuppression, secondary MDS/AML, hemorrhagic cystitis (cyclophosphamide — prevent with mesna), gonadal toxicity |
| Antimetabolites | Methotrexate, 5-FU, cytarabine, 6-MP, gemcitabine, capecitabine | Mimic nucleotides or inhibit nucleotide synthesis; S-phase specific | Mucositis, diarrhea, myelosuppression; methotrexate — renal toxicity (hydrate + leucovorin rescue); 5-FU — DPD deficiency causes fatal toxicity |
| Anthracyclines | Doxorubicin, daunorubicin, idarubicin, epirubicin | DNA intercalation, topoisomerase II inhibition, free radical generation | Dose-dependent cardiotoxicity (cumulative dose limit: doxorubicin ~450 mg/m²); dexrazoxane is cardioprotective; myelosuppression; red urine |
| Platinum agents | Cisplatin, carboplatin, oxaliplatin | DNA cross-linking | Cisplatin: nephrotoxicity (aggressive hydration), ototoxicity, peripheral neuropathy. Carboplatin: thrombocytopenia (dose by AUC/Calvert formula). Oxaliplatin: cold-induced neuropathy |
| Taxanes | Paclitaxel, docetaxel | Stabilize microtubules; M-phase specific | Peripheral neuropathy, myelosuppression (neutropenia), hypersensitivity reactions, alopecia |
| Vinca alkaloids | Vincristine, vinblastine | Inhibit microtubule assembly; M-phase specific | Vincristine: peripheral neuropathy (dose-limiting), constipation (autonomic neuropathy); IT vincristine is FATAL |
| Topoisomerase inhibitors | Etoposide (topo II), irinotecan/topotecan (topo I) | Prevent DNA religation | Etoposide: secondary AML (11q23 translocations). Irinotecan: diarrhea (early — cholinergic; late — secretory, treat with loperamide) |
Vincristine must NEVER be administered intrathecally. Accidental intrathecal injection causes ascending paralysis and death. Vincristine should be dispensed in a small-volume minibag (not a syringe) to prevent confusion with intrathecal medications. This is a never event.
33 Immunotherapy & Targeted Therapy
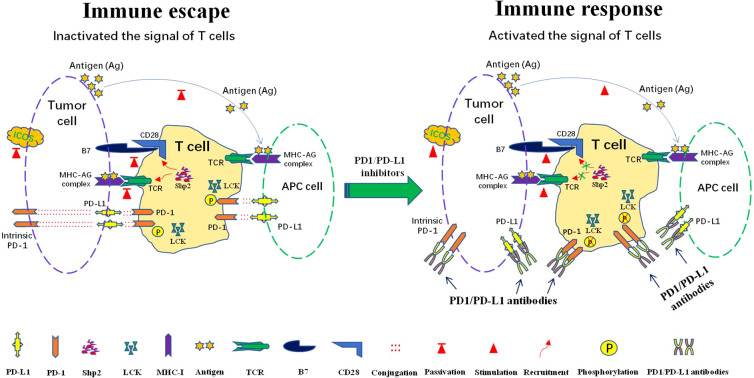
Immune Checkpoint Inhibitors
| Target | Agents | FDA-Approved Indications (Selected) |
|---|---|---|
| PD-1 | Nivolumab (Opdivo), pembrolizumab (Keytruda), cemiplimab | Melanoma, NSCLC, RCC, HL, HNSCC, urothelial, MSI-H/dMMR solid tumors (pembrolizumab), gastric, esophageal, HCC, Merkel cell |
| PD-L1 | Atezolizumab (Tecentriq), durvalumab (Imfinzi), avelumab | NSCLC, urothelial, SCLC, HCC, Merkel cell |
| CTLA-4 | Ipilimumab (Yervoy), tremelimumab | Melanoma, RCC (combination with nivolumab), mesothelioma, HCC |
Immune-Related Adverse Events (irAEs)
| Organ | irAE | Management |
|---|---|---|
| Skin | Rash, pruritus, vitiligo (favorable in melanoma) | Topical steroids; hold if severe |
| GI | Colitis, diarrhea (especially ipilimumab) | High-dose IV steroids; infliximab or vedolizumab if steroid-refractory |
| Endocrine | Hypothyroidism, hyperthyroidism, adrenal insufficiency, hypophysitis, type 1 DM | Hormone replacement (thyroid hormone, hydrocortisone); often permanent |
| Hepatic | Hepatitis (elevated transaminases) | Hold checkpoint inhibitor; high-dose steroids; mycophenolate if refractory |
| Pulmonary | Pneumonitis | Hold therapy; steroids; may need infliximab or cyclophosphamide |
| Cardiac | Myocarditis (rare but high mortality) | ICU; high-dose IV methylprednisolone; cardiology consultation |
Key Targeted Therapies
| Target | Agent Examples | Indication |
|---|---|---|
| HER2 | Trastuzumab, pertuzumab, T-DXd (ado-trastuzumab emtansine) | HER2+ breast cancer, gastric cancer |
| EGFR | Osimertinib, erlotinib, afatinib | EGFR-mutant NSCLC |
| ALK | Alectinib, lorlatinib, crizotinib | ALK-rearranged NSCLC |
| BRAF | Dabrafenib + trametinib, vemurafenib + cobimetinib, encorafenib | BRAF V600E melanoma, NSCLC, colorectal (with anti-EGFR) |
| BCR-ABL | Imatinib, dasatinib, nilotinib, ponatinib | CML, Ph+ ALL |
| VEGF/VEGFR | Bevacizumab, ramucirumab, sunitinib, sorafenib, lenvatinib, cabozantinib | RCC, HCC, CRC, glioblastoma, thyroid |
| CDK4/6 | Palbociclib, ribociclib, abemaciclib | HR+/HER2- breast cancer |
| PARP | Olaparib, niraparib, rucaparib, talazoparib | BRCA-mutant breast/ovarian/prostate/pancreatic cancer |
34 Oncologic Emergencies
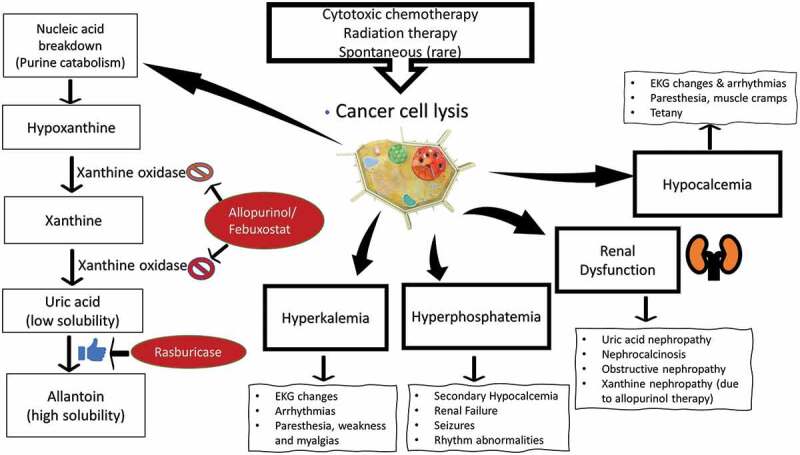
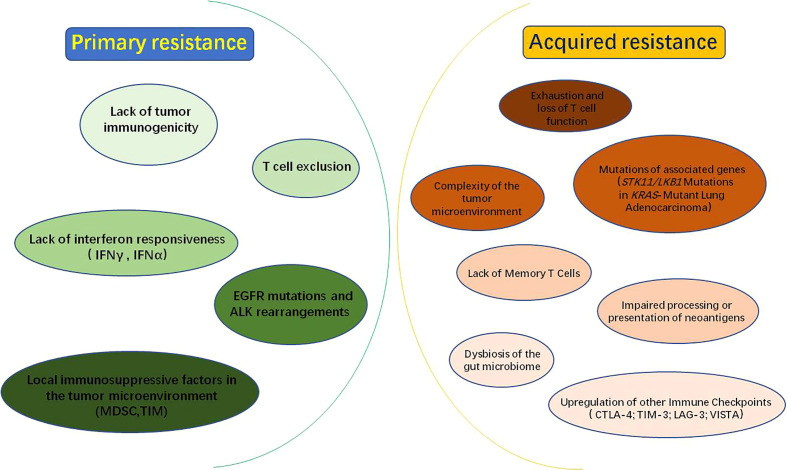
Pathophysiology: Rapid cell death releases intracellular contents: ↑ potassium, ↑ phosphate, ↑ uric acid, ↓ calcium (calcium-phosphate precipitation). Leads to acute kidney injury, cardiac arrhythmias, seizures. Most common with Burkitt lymphoma, ALL, AML with high WBC, and any bulky aggressive lymphoma.
Prevention: Aggressive IV hydration (200–250 mL/hr), allopurinol (moderate risk), rasburicase (high risk — recombinant urate oxidase, rapidly lowers uric acid; contraindicated in G6PD deficiency). Monitor labs every 6–8 hours.
Treatment: Correct hyperkalemia (insulin/glucose, kayexalate, dialysis); phosphate binders; IV calcium only for symptomatic hypocalcemia; dialysis for refractory electrolyte abnormalities or oliguria.
Definition: ANC <500/μL (or expected to decline to <500) + single temperature ≥38.3°C (101°F) or ≥38.0°C (100.4°F) sustained for ≥1 hour.
Management: Blood cultures (2 sets, peripheral + line if present) → start empiric anti-pseudomonal beta-lactam (cefepime, piperacillin-tazobactam, or meropenem) within 60 minutes. Add vancomycin only for specific indications (hemodynamic instability, suspected catheter infection, skin/soft tissue infection, MRSA risk). MASCC score stratifies low-risk (outpatient oral fluoroquinolone + amoxicillin-clavulanate) vs high-risk (inpatient IV). Add antifungal (micafungin, voriconazole) if fever persists ≥4–7 days despite antibiotics.
Mechanisms: PTHrP secretion (most common — squamous cell cancers, renal, breast), osteolytic metastases (breast, myeloma), 1,25-dihydroxyvitamin D production (lymphoma). Symptoms: confusion, constipation, polyuria, dehydration ("stones, bones, groans, and psychic moans").
Treatment: Aggressive NS hydration (200–300 mL/hr) → IV zoledronic acid 4 mg (onset 2–4 days) or denosumab (for bisphosphonate-refractory); calcitonin 4 IU/kg for rapid but transient effect (tachyphylaxis within 48 hours); treat underlying malignancy.
SVC syndrome: Obstruction of SVC (usually by mediastinal mass — lung cancer, lymphoma, or catheter-associated thrombosis). Presents with facial/upper extremity swelling, dyspnea, headache, distended neck veins. Treatment: emergent radiation or chemotherapy (if chemo-sensitive tumor like lymphoma/SCLC); stenting for rapid relief; thrombolytics if thrombosis-related.
Spinal cord compression: Oncologic emergency requiring immediate action. Presents with back pain (earliest and most common symptom), weakness, sensory level, bowel/bladder dysfunction. Dexamethasone 10 mg IV bolus immediately, then 4 mg q6h. Urgent MRI of entire spine. Radiation therapy (most common treatment), surgical decompression if single level of compression with good prognosis, or for tissue diagnosis.
35 Blood Products & Transfusion
Blood Product Indications & Thresholds
| Product | Contents | Indication / Threshold | Expected Effect |
|---|---|---|---|
| Packed RBCs (pRBCs) | RBCs with reduced plasma; ~250 mL/unit; Hct ~55–65% | Hb <7 g/dL (restrictive, most patients per TRICC trial); <8 g/dL (cardiac disease, acute coronary syndrome); symptomatic anemia at any level | 1 unit raises Hb ~1 g/dL |
| Platelets | Single donor apheresis or pooled (4–6 random donor units) | <10K (prophylactic); <50K (active bleeding or pre-procedure); <100K (neurosurgery/ocular surgery) | 1 apheresis unit raises count ~30–50K |
| FFP (Fresh Frozen Plasma) | All coagulation factors; ~250 mL/unit | Active bleeding + INR >1.5; massive transfusion; TTP (substrate for plasma exchange); DIC with bleeding | 1 unit raises factors ~3–5% |
| Cryoprecipitate | Fibrinogen, factor VIII, vWF, factor XIII, fibronectin | Fibrinogen <100–150 mg/dL (especially in DIC, massive transfusion); hemophilia A / vWD (if specific concentrates unavailable) | 1 pool (10 units) raises fibrinogen ~60–80 mg/dL |
Special Processing
| Modification | Purpose | Indications |
|---|---|---|
| Leukoreduction | Removes WBCs to prevent febrile reactions, CMV transmission, HLA alloimmunization | Universal in most blood banks; mandatory for chronically transfused patients |
| Irradiation | Prevents transfusion-associated GVHD by inactivating donor T lymphocytes | Immunocompromised patients (SCT recipients, Hodgkin lymphoma, purine analog therapy, congenital immunodeficiency), HLA-matched/directed donor products |
| CMV-negative | Reduces CMV transmission risk | CMV-negative transplant recipients with CMV-negative donors; neonates |
| Washed | Removes plasma proteins (including IgA) | IgA-deficient patients with anti-IgA antibodies (anaphylaxis risk); severe allergic transfusion reactions |
Massive Transfusion Protocol
Defined as transfusion of ≥10 units pRBCs in 24 hours or ≥4 units in 1 hour with ongoing bleeding. Protocol: 1:1:1 ratio of pRBCs : FFP : platelets (based on PROPPR trial). Administer cryoprecipitate to maintain fibrinogen >150 mg/dL. Consider tranexamic acid (TXA) within 3 hours of injury (CRASH-2 trial). Monitor for and treat hypothermia, hypocalcemia (citrate toxicity from transfused products), and acidosis.
36 Transfusion Reactions
| Reaction | Timing | Mechanism | Signs/Symptoms | Management |
|---|---|---|---|---|
| Acute hemolytic | Minutes to hours | ABO incompatibility (clerical error); preformed recipient antibodies lyse donor RBCs | Fever, rigors, flank pain, dark urine (hemoglobinuria), hypotension, DIC | STOP transfusion immediately; aggressive IV fluids to maintain renal perfusion; send direct Coombs, repeat type and crossmatch; supportive care for DIC |
| Febrile non-hemolytic (FNHTR) | During or within 4 hours | Cytokines from stored WBCs or recipient antibodies to donor WBC antigens | Temperature rise ≥1°C, rigors; no hemolysis | Stop transfusion; antipyretics; rule out hemolytic reaction; use leukoreduced products in future |
| Allergic (minor) | During transfusion | Recipient IgE against donor plasma proteins | Urticaria, pruritus, flushing | Pause transfusion; diphenhydramine; can restart if symptoms resolve |
| Anaphylactic | After small volume infused | Anti-IgA antibodies in IgA-deficient recipients | Hypotension, bronchospasm, angioedema, no fever | Stop transfusion; epinephrine; future: use washed or IgA-deficient products |
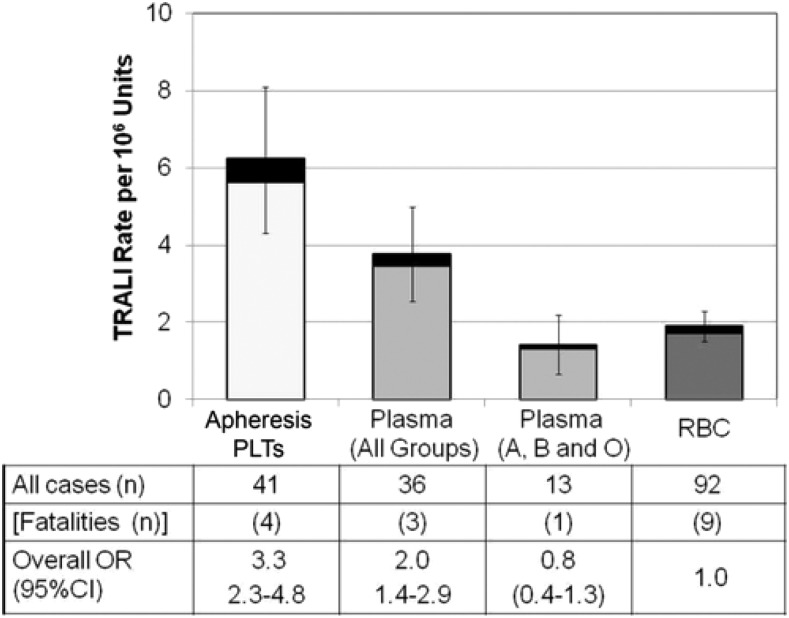
| TRALI | Within 6 hours | Donor anti-HLA/anti-neutrophil antibodies activate recipient neutrophils in pulmonary vasculature | Acute respiratory distress, bilateral infiltrates on CXR, hypoxia, NO volume overload | Stop transfusion; supportive (oxygen, may need intubation); NO diuretics (distinguish from TACO); resolves in 48–72 hours usually |
| TACO | Within 6 hours | Volume overload (not immune-mediated) | Dyspnea, hypertension, jugular venous distension, pulmonary edema on CXR, elevated BNP | Stop or slow transfusion; diuretics (furosemide); upright positioning |
| Delayed hemolytic | 3–28 days post-transfusion | Anamnestic antibody response to minor RBC antigens | Falling Hb, jaundice, positive DAT, new antibody on screen | Usually mild; supportive; future: antigen-negative blood |
| Transfusion-associated GVHD | 1–6 weeks | Donor T lymphocytes attack recipient tissues | Rash, diarrhea, liver failure, pancytopenia; >90% mortality | Prevention: irradiate products for at-risk patients; treatment largely unsuccessful |
For any suspected transfusion reaction: 1) STOP the transfusion immediately. 2) Maintain IV access with normal saline. 3) Verify patient identity and blood product labels (clerical check). 4) Send blood bank samples (post-transfusion specimen, bag with remaining product). 5) Treat supportively based on reaction type. 6) Report to blood bank.
37 Imaging in Hematology-Oncology
| Modality | Indications | Key Points |
|---|---|---|
| PET/CT (FDG) | Staging and response assessment for lymphoma (HL, DLBCL, aggressive NHL); staging solid tumors; PET-adapted therapy | Deauville 5-point scale for lymphoma response (1–3 = negative, 4–5 = positive); not useful for indolent lymphomas or CLL (variable FDG avidity) |
| Bone marrow biopsy | Diagnosis/staging of leukemias, lymphomas, myeloma, MDS, MPN, aplastic anemia, cytopenias of unknown cause | Posterior iliac crest; aspirate for morphology/flow cytometry/cytogenetics; biopsy core for architecture/fibrosis; "dry tap" suggests fibrosis (MF) or packed marrow (AML) |
| Flow cytometry | Immunophenotyping of leukemias, lymphomas, PNH, MRD detection | Identifies cell surface markers (CD antigens); essential for classifying hematologic malignancies; peripheral blood or bone marrow sample |
| Whole-body low-dose CT | Myeloma bone disease (replacing skeletal survey) | More sensitive than plain films for lytic lesions; avoids radiation of full skeletal survey series |
| MRI | Spinal cord compression, brain metastases, bone marrow infiltration, cardiac iron overload (T2*) | Most sensitive for cord compression (whole spine); T2* MRI for hepatic/cardiac iron in transfusion-dependent patients |
| Doppler ultrasound | DVT diagnosis, post-transplant hepatic veno-occlusive disease | Compression ultrasound: non-compressible vein = DVT |
38 Classification Systems
| System | Disease | Components |
|---|---|---|
| WHO Classification | All heme neoplasms | Integrates morphology, immunophenotype, genetics, clinical features; updated 5th edition 2022 |
| Ann Arbor / Lugano | Hodgkin & Non-Hodgkin lymphoma | Stages I–IV; A/B symptoms; Lugano adds PET-based response (Deauville criteria) |
| Rai / Binet | CLL | Rai: 0–IV (lymphocytosis → thrombocytopenia); Binet: A–C (nodal areas + cytopenias) |
| ISS / R-ISS | Multiple myeloma | ISS: beta-2M + albumin; R-ISS adds LDH + high-risk cytogenetics [t(4;14), t(14;16), del(17p)] |
| IPSS-R | MDS | Cytogenetics, marrow blast %, Hb, platelets, ANC → 5 risk groups |
| ELN 2022 | AML | Favorable / intermediate / adverse based on cytogenetics and molecular markers |
| DIPSS / DIPSS-Plus | Myelofibrosis | Age, symptoms, Hb, WBC, blasts, karyotype, transfusion dependence, platelets |
| IPI / FLIPI | DLBCL / Follicular lymphoma | IPI: age, stage, LDH, ECOG, extranodal sites; FLIPI: age, stage, Hb, LDH, nodal areas |
| ISTH DIC Score | DIC | Platelet count, D-dimer, PT prolongation, fibrinogen; ≥5 = overt DIC |
| 4T Score | HIT | Thrombocytopenia, timing, thrombosis, other causes; 0–3 low, 4–5 intermediate, 6–8 high |
39 Medications Master Table
Chemotherapy Agents
| Drug | Class | Key Toxicity | Notes |
|---|---|---|---|
| Doxorubicin | Anthracycline | Cardiotoxicity (cumulative) | Max ~450 mg/m²; echo monitoring |
| Cyclophosphamide | Alkylating | Hemorrhagic cystitis | Mesna prophylaxis; secondary AML |
| Cisplatin | Platinum | Nephrotoxicity, ototoxicity | Aggressive hydration essential |
| Methotrexate | Antimetabolite | Mucositis, renal, hepatic | Leucovorin rescue; alkalinize urine |
| Vincristine | Vinca alkaloid | Neuropathy | NEVER intrathecal; cap 2 mg |
| Bleomycin | Antitumor antibiotic | Pulmonary fibrosis | Cumulative dose limit; PFTs |
| Cytarabine (Ara-C) | Antimetabolite | Cerebellar toxicity (high dose) | Check cerebellar function before each dose |
Targeted & Immunotherapy Agents
| Drug | Target/Mechanism | Key Toxicity | Indication |
|---|---|---|---|
| Rituximab | Anti-CD20 | Infusion reactions, HBV reactivation | NHL, CLL, AIHA, ITP |
| Imatinib | BCR-ABL TKI | Edema, GI, myelosuppression | CML, Ph+ ALL, GIST |
| Bortezomib | Proteasome inhibitor | Peripheral neuropathy, thrombocytopenia | Myeloma, mantle cell |
| Lenalidomide | IMiD (immunomodulatory) | Myelosuppression, VTE | Myeloma, MDS del(5q), MCL |
| Venetoclax | BCL-2 inhibitor | TLS (dose ramp-up required) | CLL, AML |
| Pembrolizumab | Anti-PD-1 | irAEs (any organ) | Multiple solid tumors, HL |
| Daratumumab | Anti-CD38 | Infusion reactions, interferes with crossmatch | Myeloma, AL amyloidosis |
| Ibrutinib | BTK inhibitor | AFib, bleeding, hypertension | CLL, MCL, WM |
Anticoagulants & Reversal Agents
| Anticoagulant | Reversal Agent | Onset of Reversal |
|---|---|---|
| UFH | Protamine sulfate | Immediate |
| LMWH | Protamine (partial reversal ~60%) | Immediate (partial) |
| Warfarin | Vitamin K + 4-factor PCC (Kcentra) for urgent; FFP if PCC unavailable | PCC: minutes; Vitamin K: 6–24 hours |
| Dabigatran | Idarucizumab (Praxbind) | Minutes |
| Rivaroxaban / Apixaban / Edoxaban | Andexanet alfa (Andexxa); 4-factor PCC (off-label) | Minutes |
Growth Factors
| Agent | Target | Indication |
|---|---|---|
| Filgrastim / pegfilgrastim (G-CSF) | Neutrophils | Chemotherapy-induced neutropenia prophylaxis; stem cell mobilization |
| Epoetin alfa / darbepoetin (EPO) | Erythrocytes | Anemia of CKD, MDS (select), chemo-induced anemia (use cautiously — thrombosis risk) |
| Romiplostim / eltrombopag (TPO agonists) | Platelets | Chronic ITP, aplastic anemia (eltrombopag) |
40 Abbreviations Master List
41 Key Trials & Treatment Protocols
| Trial | Disease | Key Finding |
|---|---|---|
| IRIS (2003) | CML | Imatinib superior to IFN-alpha + cytarabine; established TKI era for CML |
| VIALE-A (2020) | AML (unfit) | Venetoclax + azacitidine improved OS vs azacitidine alone (14.7 vs 9.6 months); new standard of care for unfit AML |
| ECHELON-1 (2018) | Hodgkin lymphoma | BV-AVD superior to ABVD in advanced HL (modified PFS benefit) |
| POLARIX (2022) | DLBCL | Pola-R-CHP showed PFS benefit over R-CHOP in previously untreated DLBCL |
| ZUMA-7 (2022) | DLBCL | Axicabtagene ciloleucel (CAR-T) superior to standard salvage chemo + auto-SCT in second-line R/R DLBCL (EFS benefit) |
| MAIA (2019) | Multiple myeloma | DRd (daratumumab-lenalidomide-dex) superior to Rd for transplant-ineligible myeloma (PFS) |
| PERSEUS (2023) | Multiple myeloma | Daratumumab-VRd + auto-SCT + dara-lenalidomide maintenance superior to VRd in transplant-eligible myeloma |
| COMFORT-I/II (2012) | Myelofibrosis | Ruxolitinib reduced spleen volume and improved symptoms vs placebo/BAT; survival benefit |
| CYTO-PV (2013) | Polycythemia vera | Hematocrit target <45% reduced cardiovascular events and death vs 45–50% |
| ECLAP (2004) | Polycythemia vera | Low-dose aspirin reduced thrombotic events without significant increase in bleeding |
| TRICC (1999) | Transfusion medicine | Restrictive transfusion (Hb <7 g/dL) non-inferior to liberal (Hb <10 g/dL) in critically ill; reduced transfusion use |
| PROPPR (2015) | Massive transfusion | 1:1:1 (pRBC:FFP:platelets) ratio improved hemostasis and reduced 24-hour mortality vs 1:1:2 |
| CRASH-2 (2010) | Trauma / bleeding | Tranexamic acid (TXA) within 3 hours of injury reduced death from hemorrhage; no benefit after 3 hours |
| TRAPS (2018) | Antiphospholipid syndrome | Rivaroxaban inferior to warfarin in triple-positive APS; DOACs not recommended for APS |
| RESONATE-2 (2015) | CLL | Ibrutinib superior to chlorambucil as first-line for elderly CLL (PFS, OS benefit) |
| ALPINE (2023) | CLL | Zanubrutinib superior to ibrutinib in R/R CLL (ORR, fewer cardiac adverse events) |
References
- Kaushansky K, et al. Williams Hematology, 10th ed. McGraw-Hill, 2021.
- Hoffman R, et al. Hematology: Basic Principles and Practice, 8th ed. Elsevier, 2023.
- NCCN Clinical Practice Guidelines in Oncology. National Comprehensive Cancer Network, 2024.
- Döhner H, et al. Diagnosis and management of AML in adults: 2022 ELN recommendations. Blood. 2022;140(12):1345–1377.
- Hallek M, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745–2760.
- Rajkumar SV, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–e548.
- Greenberg PL, et al. Revised International Prognostic Scoring System for myelodysplastic syndromes. Blood. 2012;120(12):2454–2465.
- Marchioli R, et al (CYTO-PV). Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22–33.
- Cuker A, et al. American Society of Hematology 2018 guidelines for management of venous thromboembolism: heparin-induced thrombocytopenia. Blood Adv. 2018;2(22):3360–3392.
- Kearon C, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest. 2016;149(2):315–352.
- Carson JL, et al (TRICC). Liberal or restrictive transfusion in high-risk patients after hip surgery. N Engl J Med. 2011;365:2453–2462.
- Holcomb JB, et al (PROPPR). Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio. JAMA. 2015;313(5):471–482.
- DiNardo CD, et al (VIALE-A). Azacitidine with venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617–629.
- Connors JM, et al (ECHELON-1). Brentuximab vedotin with chemotherapy for stage III or IV Hodgkin's lymphoma. N Engl J Med. 2018;378(4):331–344.
- Tilly H, et al (POLARIX). Polatuzumab vedotin in previously untreated diffuse large B-cell lymphoma. N Engl J Med. 2022;386(4):351–363.
- Facon T, et al (MAIA). Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. 2019;380(22):2104–2115.
- Postow MA, et al. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. 2018;378(2):158–168.
- Howard SC, et al. The tumor lysis syndrome. N Engl J Med. 2011;364(19):1844–1854.
- Swerdlow SH, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised 5th ed. IARC, 2022.