Medical Genetics
Every genetic syndrome, inheritance pattern, chromosomal disorder, inborn error of metabolism, cancer predisposition syndrome, genetic test, prenatal screening modality, classification system, and therapeutic strategy in one place.
01 Molecular Genetics Essentials
Medical genetics is the branch of medicine dealing with the diagnosis and management of hereditary disorders. A firm understanding of molecular biology — from DNA structure through protein synthesis — is the foundation upon which all clinical genetics rests. The human genome contains approximately 3.2 billion base pairs distributed across 23 pairs of chromosomes (22 autosomal pairs and 1 pair of sex chromosomes), encoding roughly 20,000–25,000 protein-coding genes.
DNA Structure & Replication
Deoxyribonucleic acid (DNA) is a double-stranded helix composed of nucleotide subunits, each consisting of a deoxyribose sugar, a phosphate group, and one of four nitrogenous bases: adenine (A), thymine (T), guanine (G), and cytosine (C). Base pairing follows Watson-Crick rules: A pairs with T (2 hydrogen bonds), G pairs with C (3 hydrogen bonds). The two strands run antiparallel (5′→3′ and 3′→5′). DNA replication is semiconservative — each daughter molecule contains one original and one newly synthesized strand. Key enzymes include helicase (unwinds), primase (RNA primer), DNA polymerase III (synthesis, 5′→3′ only), and DNA ligase (joins Okazaki fragments on the lagging strand).
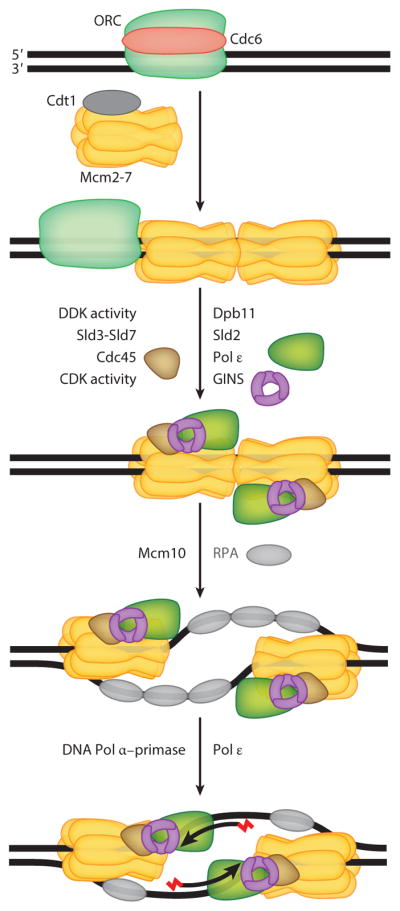
Gene Structure
A typical human gene consists of: promoter (upstream regulatory region, often containing a TATA box ~25 bp upstream of the transcription start site), exons (protein-coding sequences that are retained in mature mRNA), introns (intervening non-coding sequences removed by splicing), and 3′ untranslated region (UTR) with a polyadenylation signal. Enhancers and silencers may be located thousands of base pairs away and regulate transcription by looping to interact with the promoter complex. The average human gene has ~8 exons, but individual genes vary enormously — the dystrophin gene (DMD) has 79 exons spanning 2.4 Mb, making it the largest known human gene.
Transcription & Translation
Transcription occurs in the nucleus: RNA polymerase II synthesizes a pre-mRNA complementary to the template (antisense) DNA strand, reading 3′→5′ and synthesizing 5′→3′. Post-transcriptional modifications include 5′ capping (7-methylguanosine), splicing (removal of introns by the spliceosome at GT-AG splice sites), and 3′ polyadenylation. The mature mRNA is exported to the cytoplasm for translation by ribosomes. Translation proceeds in codons (triplets of nucleotides); the start codon is AUG (methionine), and the three stop codons are UAA, UAG, UGA. Transfer RNA (tRNA) molecules carry specific amino acids matched to codons via their anticodon loops.
Types of Mutations
| Mutation Type | Definition | Example |
|---|---|---|
| Missense | Single nucleotide change altering one amino acid | HbS (Glu6Val in β-globin) |
| Nonsense | Creates a premature stop codon | Many CF mutations |
| Frameshift | Insertion or deletion not a multiple of 3, altering reading frame | Duchenne dystrophin deletions |
| In-frame deletion/insertion | Loss or gain of codon(s) without disrupting reading frame | F508del in CFTR (loss of Phe508) |
| Splice site | Disruption of GT/AG donor/acceptor sites | Some β-thalassemia mutations |
| Trinucleotide repeat expansion | Unstable expansion of 3-nucleotide repeats | CAG in Huntington disease |
| Promoter/regulatory | Altered gene expression without coding change | Some Gilbert syndrome variants |
02 Chromosome Structure & Karyotype Notation
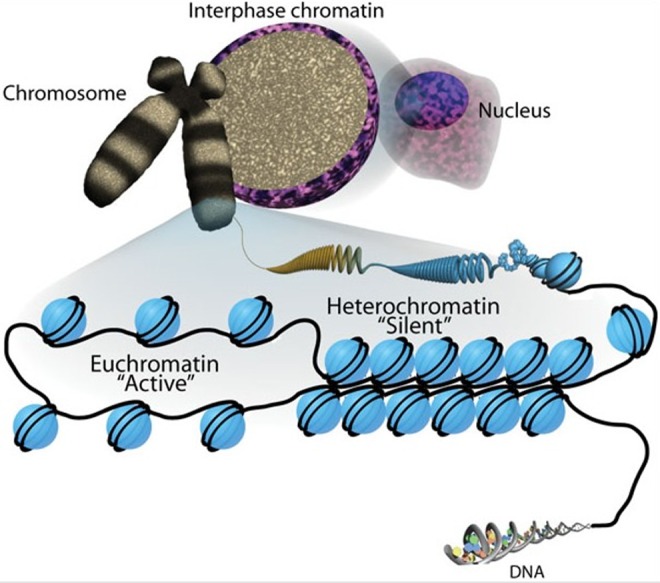
Humans have 46 chromosomes: 22 pairs of autosomes (numbered 1–22 by size) and one pair of sex chromosomes (XX in females, XY in males). Each chromosome consists of a single, continuous DNA molecule wrapped around histone proteins forming nucleosomes, which further compact into chromatin and ultimately the visible chromosome during cell division.
Chromosome Architecture
Key structural landmarks: the centromere divides the chromosome into a short arm (p, for "petit") and a long arm (q). Based on centromere position, chromosomes are classified as metacentric (centromere near center — chromosomes 1, 3, 16, 19, 20), submetacentric (centromere off-center — most chromosomes), or acrocentric (centromere near the end — chromosomes 13, 14, 15, 21, 22). The telomeres are repetitive TTAGGG sequences at chromosome ends that protect against degradation and shorten with each cell division (relevant to aging and cancer biology). Acrocentric chromosomes have satellite stalks containing ribosomal RNA genes — these are clinically significant because they can participate in Robertsonian translocations.
Karyotype Notation (ISCN)
Karyotypes follow the International System for Human Cytogenomic Nomenclature (ISCN). The standard format is: total chromosome number, sex chromosomes, abnormality.
| Karyotype | Interpretation |
|---|---|
| 46,XX | Normal female |
| 46,XY | Normal male |
| 47,XX,+21 | Female with trisomy 21 (Down syndrome) |
| 47,XY,+18 | Male with trisomy 18 (Edwards syndrome) |
| 45,X | Turner syndrome |
| 47,XXY | Klinefelter syndrome |
| 46,XX,del(5)(p15.2) | Female with deletion of 5p15.2 (Cri-du-chat) |
| 45,XY,rob(14;21)(q10;q10) | Male with Robertsonian translocation 14;21 |
| 46,XY,t(9;22)(q34;q11.2) | Male with reciprocal translocation (Philadelphia chromosome) |
| 46,XX,inv(9)(p11q13) | Female with pericentric inversion of chromosome 9 (common variant) |
| 47,XXX | Triple X syndrome |
| 47,XYY | 47,XYY syndrome |
| 46,XX/47,XX,+21 | Mosaic Down syndrome (two cell lines) |
| 69,XXX | Triploidy |
del = deletion; dup = duplication; inv = inversion; t = translocation; rob = Robertsonian translocation; ins = insertion; r = ring chromosome; i = isochromosome; mar = marker chromosome; + = gain; − = loss; p = short arm; q = long arm; mos = mosaic.
Cell Division
Mitosis produces two genetically identical diploid daughter cells (for somatic growth and repair). Meiosis produces four haploid gametes and is unique for two features critical to genetic diversity: crossing over (homologous recombination during meiosis I) and independent assortment of homologous chromosomes. Errors in meiosis cause nondisjunction — failure of homologs (meiosis I) or sister chromatids (meiosis II) to separate — resulting in aneuploidy (trisomy or monosomy) in the offspring. Nondisjunction in meiosis I produces gametes with both homologs (one disomic, one nullisomic); nondisjunction in meiosis II produces one disomic gamete with identical copies, one nullisomic, and two normal.
03 Inheritance Patterns
Understanding the patterns by which genetic traits are transmitted through families is essential for genetic counseling, risk assessment, and family planning. The major Mendelian and non-Mendelian patterns are described below.
Autosomal Dominant (AD)
Only one mutant allele needed for disease expression. Each affected individual has a 50% chance of passing the mutation to each offspring, regardless of sex. Key features: vertical transmission (appears in every generation), male-to-male transmission possible (distinguishes from X-linked), variable expressivity (different features/severity among carriers of same mutation), reduced penetrance (some carriers never manifest disease). New mutations (de novo) are common, especially for conditions reducing reproductive fitness (e.g., achondroplasia — ~80% de novo). Examples: Marfan syndrome, neurofibromatosis type 1, Huntington disease, familial hypercholesterolemia, ADPKD.
Autosomal Recessive (AR)
Two mutant alleles required (homozygous or compound heterozygous). Carrier parents (heterozygous) have a 25% risk per pregnancy of an affected child, 50% carriers, 25% unaffected non-carriers. Key features: horizontal pattern (siblings affected, parents unaffected), increased incidence with consanguinity, often involves enzyme deficiencies (loss of function). Examples: cystic fibrosis, sickle cell disease, PKU, Tay-Sachs, spinal muscular atrophy.
X-Linked Recessive (XLR)
Gene on X chromosome; primarily males affected (hemizygous). Carrier females are usually unaffected but may show mild manifestations due to skewed X-inactivation. Key features: no male-to-male transmission (father passes Y to sons), affected males related through carrier females, all daughters of affected males are obligate carriers. Carrier mother has 50% chance of affected sons and 50% chance of carrier daughters. Examples: Duchenne muscular dystrophy, hemophilia A and B, G6PD deficiency, Fabry disease.
X-Linked Dominant (XLD)
Affects both males and females with one mutant allele on X, but often more severe or lethal in males. Affected females transmit to 50% of sons and 50% of daughters; affected males transmit to all daughters and no sons. Examples: Rett syndrome (MECP2 — almost exclusively in females; lethal in most males), Incontinentia pigmenti (IKBKG), X-linked hypophosphatemic rickets (PHEX).
Mitochondrial Inheritance
Mitochondrial DNA (mtDNA) is a 16.5 kb circular molecule encoding 37 genes (13 polypeptides, 22 tRNAs, 2 rRNAs). Inheritance is strictly maternal — mitochondria in the oocyte are passed to all offspring, but an affected father cannot transmit. Heteroplasmy (mixture of normal and mutant mtDNA within a cell) explains variable expressivity and the threshold effect — disease manifests when the proportion of mutant mtDNA exceeds a tissue-specific threshold. Examples: MELAS, MERRF, Leber hereditary optic neuropathy.
Multifactorial (Complex) Inheritance
Caused by the interaction of multiple genes and environmental factors. Does not follow simple Mendelian patterns. Recurrence risks are empiric (based on population data) rather than calculated. Features: risk increases with number of affected relatives, severity of disease, and relatedness of affected individuals. Examples: neural tube defects, cleft lip/palate, congenital heart defects, type 2 diabetes, hypertension.
04 Genetic Variation & Epigenetics
Types of Genetic Variation
| Variant Type | Definition | Clinical Significance |
|---|---|---|
| SNP (single nucleotide polymorphism) | Single base change present in ≥1% of population | Pharmacogenomics, GWAS risk loci |
| SNV (single nucleotide variant) | Single base change at any frequency | May be pathogenic or benign |
| CNV (copy number variant) | Gain or loss of DNA segments ≥1 kb | Microdeletion/microduplication syndromes |
| Indel | Small insertion or deletion (<1 kb) | Frameshifts, in-frame changes |
| Trinucleotide repeat expansion | Unstable expansion of 3-base repeat units | Huntington, Fragile X, myotonic dystrophy |
| Structural variant | Large-scale rearrangement (>50 bp) | Translocations, inversions, complex rearrangements |
Epigenetics
Heritable changes in gene expression without alteration of DNA sequence. Major epigenetic mechanisms include:
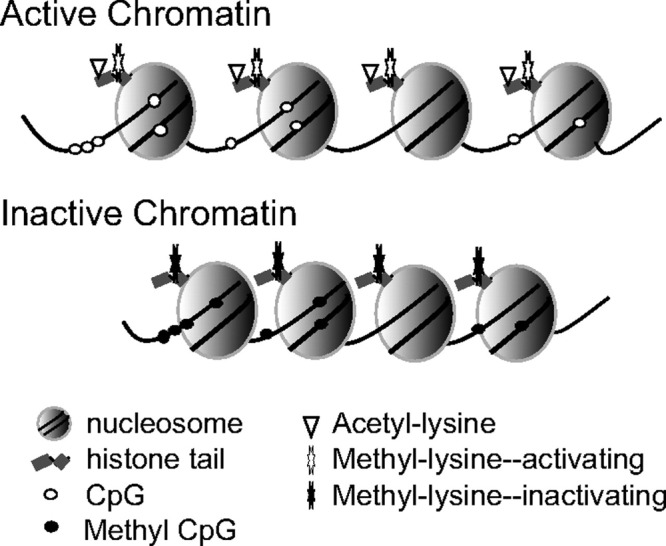
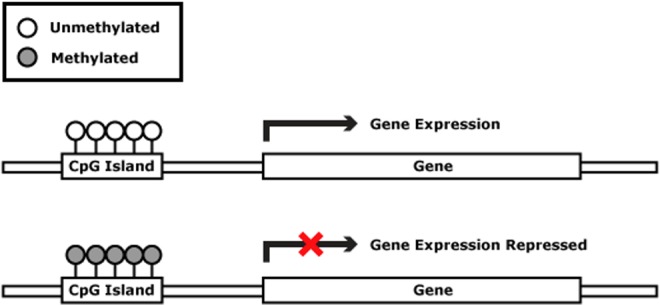
DNA methylation: Addition of a methyl group to cytosine at CpG dinucleotides by DNA methyltransferases (DNMTs). Promoter methylation typically silences gene expression. Aberrant methylation is implicated in cancer (tumor suppressor silencing) and imprinting disorders.
Histone modification: Post-translational modifications of histone tails (acetylation, methylation, phosphorylation, ubiquitination) that alter chromatin structure. Histone acetylation generally opens chromatin (euchromatin, active transcription); deacetylation compacts chromatin (heterochromatin, gene silencing).
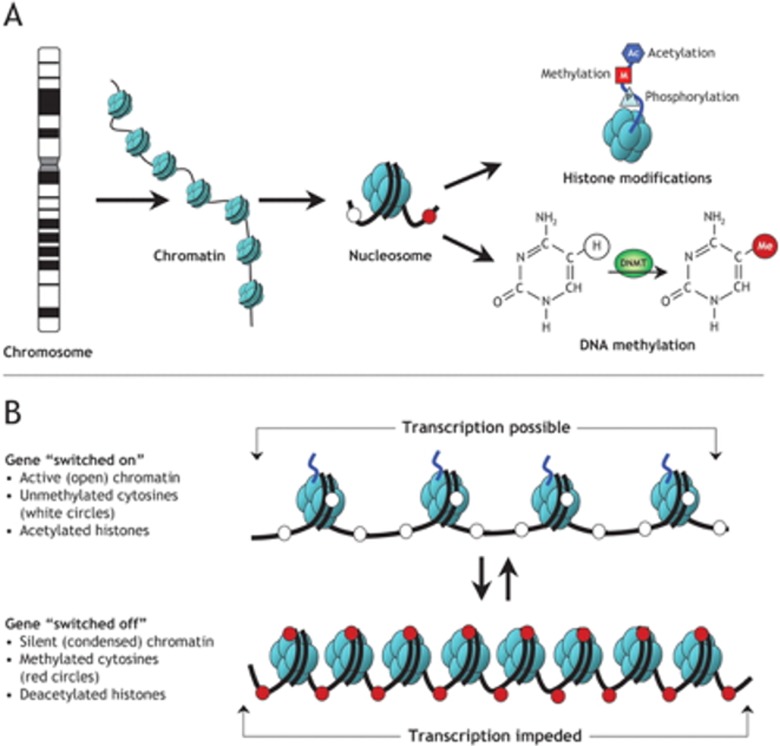
Genomic imprinting: Parent-of-origin-specific gene expression. Some genes are expressed only from the maternally inherited allele, others only from the paternally inherited allele. Mediated by differential methylation established in gametogenesis. Clinically critical examples:
- Prader-Willi syndrome: loss of paternal 15q11-13 expression (paternal deletion, maternal UPD, or imprinting center defect)
- Angelman syndrome: loss of maternal UBE3A expression at 15q11-13 (maternal deletion, paternal UPD, UBE3A mutation, or imprinting defect)
- Beckwith-Wiedemann syndrome: overgrowth disorder at 11p15.5 involving IGF2/H19
X-inactivation (Lyonization): Random inactivation of one X chromosome in each cell of 46,XX females, occurring early in embryogenesis (~day 16). Mediated by the XIST gene (X-inactivation specific transcript) on Xq13. Results in dosage compensation between males (1 active X) and females (1 active X, 1 Barr body). X-inactivation is random but fixed — all daughter cells maintain the same inactive X. Skewed X-inactivation (>80:20 ratio) can cause carrier females to manifest X-linked recessive conditions (e.g., a carrier female with hemophilia A symptoms).
05 Key Terminology & Abbreviations
| Term | Definition |
|---|---|
| Allele | One of two or more versions of a gene at a given locus |
| Penetrance | Proportion of individuals with a pathogenic variant who manifest disease; complete = 100%, reduced <100% |
| Expressivity | Variation in clinical features among individuals with the same genotype |
| Pleiotropy | A single gene affecting multiple organ systems (e.g., Marfan — eyes, heart, skeleton) |
| Genetic heterogeneity | Same phenotype caused by mutations in different genes (locus heterogeneity) or different mutations in the same gene (allelic heterogeneity) |
| Anticipation | Earlier onset or more severe disease in successive generations; characteristic of trinucleotide repeat disorders |
| Compound heterozygote | Two different pathogenic variants in the same gene (one on each allele) |
| Hemizygous | Only one copy of a gene (e.g., X-linked genes in males) |
| Proband / Index case | The individual through whom a family with a genetic disorder is first identified |
| Consanguinity | Mating between related individuals; increases risk of AR disorders |
| De novo | New mutation arising in the proband, not inherited from either parent |
| Mosaicism | Presence of two or more genetically distinct cell lines in one individual |
| Gonadal mosaicism | Mutation present in a proportion of germ cells but not somatic cells; explains recurrence in siblings with unaffected parents |
| Loss of heterozygosity (LOH) | Loss of the normal allele in a cell already carrying one pathogenic variant; key in tumor suppressor gene inactivation |
| Haploinsufficiency | One functional copy of a gene insufficient for normal function; mechanism of many AD disorders |
| Dominant negative | Mutant protein interferes with the normal protein product (e.g., collagen disorders) |
| Gain of function | Mutation confers new or enhanced protein activity (e.g., achondroplasia FGFR3) |
06 Autosomal Trisomies (21, 18, 13)
Down Syndrome (Trisomy 21)
The most common autosomal aneuploidy compatible with long-term survival, affecting ~1 in 700 live births. Three cytogenetic types:
| Type | Frequency | Karyotype | Recurrence Risk |
|---|---|---|---|
| Free trisomy 21 | ~95% | 47,XX,+21 or 47,XY,+21 | ~1% or maternal age-related risk (whichever higher) |
| Robertsonian translocation | ~3–4% | 46,XX,rob(14;21)(q10;q10),+21 | If parent is carrier: ~10–15% (mother) or ~3–5% (father); if rob(21;21) → 100% |
| Mosaic | ~1–2% | 46,XX/47,XX,+21 | Low (<1%); phenotype often milder |
Clinical features: characteristic facies (upslanting palpebral fissures, epicanthal folds, flat nasal bridge, small ears, protruding tongue, Brushfield spots on iris), hypotonia, intellectual disability (mild to moderate, mean IQ ~50), single transverse palmar crease, sandal gap (wide space between 1st and 2nd toes), short stature, brachycephaly.
Complications requiring surveillance:
- Cardiac: congenital heart defects in ~40–50% — most common is endocardial cushion defect (AVSD), followed by VSD, ASD, tetralogy of Fallot
- GI: duodenal atresia ("double bubble" sign), Hirschsprung disease, celiac disease
- Hematologic: transient myeloproliferative disorder (TMD) in neonates (~10%), increased risk of ALL (10–20×) and AML (150× for AMKL)
- Endocrine: hypothyroidism (~15–20%, annual screening), type 1 diabetes
- Neurologic: early-onset Alzheimer disease (nearly universal neuropathology by age 40, clinical dementia in ~50–70% by age 60 — due to APP gene on chromosome 21)
- Cervical spine: atlantoaxial instability (~15%), requires screening before anesthesia/sports
- ENT: hearing loss (75%), obstructive sleep apnea (50–75%)
- Ophthalmologic: refractive errors, strabismus, cataracts
Edwards Syndrome (Trisomy 18)
Second most common autosomal trisomy at birth (~1 in 5,000 live births). 95% due to free trisomy 18 from maternal meiotic nondisjunction. Clinical features: severe intellectual disability, IUGR, prominent occiput, micrognathia, clenched fists with overlapping fingers (index over 3rd, 5th over 4th), rocker-bottom feet, short sternum, congenital heart defects (~90% — VSD, PDA most common), omphalocele, renal malformations. Prognosis: median survival ~5–15 days; ~5–10% survive to 1 year.
Patau Syndrome (Trisomy 13)
Third most common autosomal trisomy (~1 in 10,000–20,000 live births). Clinical features: severe intellectual disability, holoprosencephaly (failure of forebrain to divide — spectrum from alobar to lobar), midline facial defects (cleft lip/palate, cyclopia in severe cases), microphthalmia, polydactyly (postaxial), cutis aplasia (scalp defects), congenital heart defects (~80%), renal anomalies. Prognosis: median survival ~7–10 days; ~5% survive to 6 months.
| Feature | Trisomy 21 | Trisomy 18 | Trisomy 13 |
|---|---|---|---|
| Hands | Single palmar crease, clinodactyly | Clenched, overlapping fingers | Polydactyly |
| Heart defect | AVSD | VSD, PDA | VSD, PDA, dextrocardia |
| Head | Brachycephaly, flat face | Prominent occiput | Holoprosencephaly, microcephaly |
| Feet | Sandal gap | Rocker-bottom | Rocker-bottom |
| Survival | Median ~60 years | Median ~5–15 days | Median ~7–10 days |
07 Sex Chromosome Disorders
Turner Syndrome (45,X)
Occurs in ~1 in 2,000–2,500 live female births. Most common sex chromosome abnormality in females. ~99% of 45,X conceptions result in spontaneous abortion (most common chromosomal cause of first-trimester miscarriage). Approximately 50% have 45,X; others are mosaic (45,X/46,XX) or have structural X abnormalities (isochromosome Xq, ring X, Xp deletion).
Clinical features: short stature (mean adult height ~147 cm without treatment — related to SHOX gene haploinsufficiency on Xp), gonadal dysgenesis (streak gonads, primary amenorrhea, infertility in most), lymphedema (especially hands and feet in neonates), webbed neck (pterygium colli, from fetal cystic hygroma resolution), shield chest with widely spaced nipples, low posterior hairline, cubitus valgus. Intelligence is typically normal, though nonverbal learning disabilities and difficulties with visuospatial processing are common.
Management:
- Growth hormone: recombinant hGH starting early childhood (can add ~5–8 cm to final height)
- Estrogen replacement: start at ~11–12 years to induce puberty; followed by combined estrogen-progesterone for uterine protection
- Cardiac screening: bicuspid aortic valve (~30%), coarctation of the aorta (~10%), aortic root dilation with risk of dissection — baseline and periodic echocardiography and cardiac MRI
- Renal: horseshoe kidney (~30%), collect system anomalies — renal ultrasound at diagnosis
- Thyroid: autoimmune thyroiditis (~25–30%) — annual screening
- Hearing: sensorineural or conductive hearing loss — audiologic monitoring
- Fertility: oocyte/embryo cryopreservation in mosaic patients with residual ovarian function; donor oocyte IVF
Aortic dissection in Turner syndrome: Risk is 100× that of the general population due to underlying aortopathy. Patients with bicuspid aortic valve, coarctation, or hypertension are at highest risk. Aortic size index (ASI) >2.5 cm/m² should prompt surgical evaluation. Any acute chest/back pain requires urgent aortic imaging.
Klinefelter Syndrome (47,XXY)
Most common sex chromosome aneuploidy in males (~1 in 600–1,000 live male births). Often underdiagnosed; only ~25% diagnosed in lifetime. Clinical features: tall stature with long legs (eunuchoid proportions), small firm testes (<4 mL), infertility (azoospermia in ~95%; some oligospermia in mosaics), gynecomastia (~40%), reduced facial/body hair, increased risk of metabolic syndrome, osteoporosis, and breast cancer (20× general male population). Intelligence is usually normal but average IQ ~10 points lower than siblings; language and learning disabilities common. Management: testosterone replacement starting at puberty; fertility options include micro-TESE with ICSI in select patients.
Other Sex Chromosome Variants
47,XYY (~1 in 1,000 males): tall stature, usually normal phenotype, normal fertility, possible learning difficulties; previously controversially associated with aggressive behavior (not supported by evidence). 47,XXX (Triple X, ~1 in 1,000 females): tall stature, usually normal phenotype and fertility, mild learning difficulties possible; most never diagnosed. 48,XXXY and 49,XXXXY: increasingly severe phenotype with additional X chromosomes (more severe intellectual disability, skeletal anomalies, hypogonadism).
08 Microdeletion & Microduplication Syndromes
Microdeletion syndromes result from submicroscopic chromosomal deletions (typically 1–5 Mb) detectable by FISH or chromosomal microarray but often below the resolution of standard G-banded karyotype (~5–10 Mb). Most occur de novo.
| Syndrome | Locus | Key Features | Key Genes |
|---|---|---|---|
| 22q11.2 deletion (DiGeorge / Velocardiofacial) | 22q11.2 | Conotruncal cardiac defects (tetralogy of Fallot, interrupted aortic arch, truncus arteriosus), thymic hypoplasia (T-cell immunodeficiency), hypocalcemia (hypoparathyroidism), palatal anomalies (velopharyngeal insufficiency, cleft palate), characteristic facies, learning disabilities, psychiatric disorders (~25% develop schizophrenia). Mnemonic: CATCH-22 (Cardiac, Abnormal facies, Thymic hypoplasia, Cleft palate, Hypocalcemia — chromosome 22) | TBX1 |
| Williams syndrome | 7q11.23 | "Cocktail party" personality (overfriendly, strong verbal skills), supravalvular aortic stenosis, elfin facies, hypercalcemia (infantile), intellectual disability, stellate iris pattern, connective tissue abnormalities | ELN (elastin) |
| Prader-Willi syndrome | 15q11-13 (paternal) | Neonatal hypotonia and feeding difficulties → hyperphagia and obesity (usually by age 2–6), short stature, hypogonadism, small hands/feet, intellectual disability (mild-moderate), behavioral problems (tantrums, OCD) | SNRPN, NDN (paternal expression) |
| Angelman syndrome | 15q11-13 (maternal) | "Happy puppet" — severe intellectual disability, absent speech, ataxic gait, seizures, frequent laughter/smiling, microcephaly, characteristic EEG (large-amplitude slow spike-wave) | UBE3A (maternal expression) |
| Smith-Magenis syndrome | 17p11.2 | Intellectual disability, self-injurious behavior (self-hugging, onychotillomania), sleep disturbance (inverted melatonin rhythm), brachycephaly, midface hypoplasia | RAI1 |
| Wolf-Hirschhorn syndrome | 4p16.3 | "Greek warrior helmet" facies (prominent glabella, broad nasal bridge), severe intellectual disability, seizures, growth retardation, cardiac defects, renal anomalies | WHSC1, LETM1 |
| Cri-du-chat syndrome | 5p15.2 | High-pitched mewing cry (due to laryngeal abnormality — resolves with age), microcephaly, round face, hypertelorism, intellectual disability, cardiac defects | CTNND2, TERT |
| 1p36 deletion syndrome | 1p36 | Intellectual disability, seizures, hearing impairment, cardiomyopathy, characteristic facies (straight eyebrows, deep-set eyes, midface hypoplasia) | Multiple |
09 Connective Tissue Disorders (Marfan, EDS)
Marfan Syndrome
Autosomal dominant disorder of connective tissue caused by mutations in FBN1 (fibrillin-1) on chromosome 15q21.1. Prevalence ~1 in 5,000. ~25% de novo mutations. Fibrillin-1 is a major component of extracellular microfibrils; its deficiency leads to dysregulated TGF-β signaling.
Revised Ghent Nosology (2010) for diagnosis — two main criteria:
- Aortic root dilation/dissection (Z-score ≥2, or aortic root dissection)
- Ectopia lentis (lens subluxation, typically superotemporal)
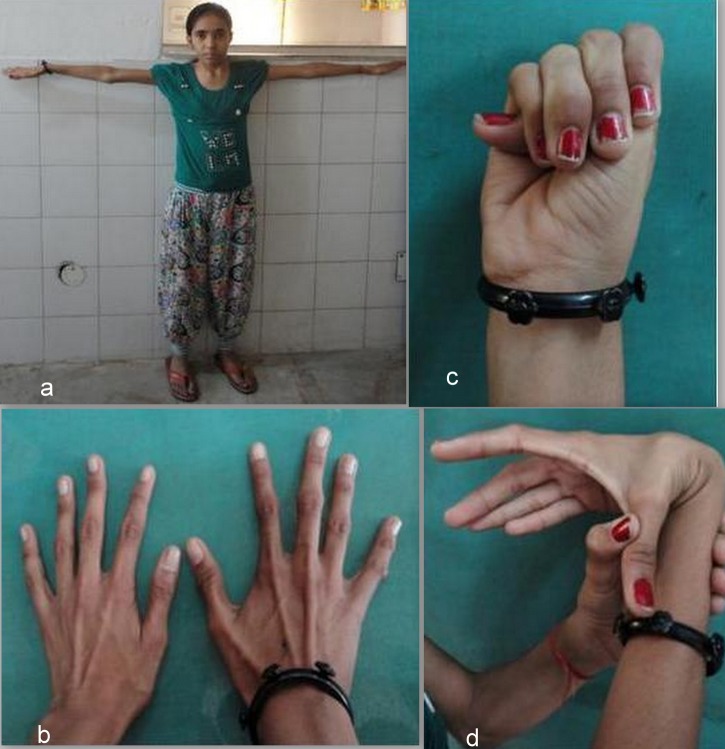
Diagnosis requires: aortic root dilation/dissection AND ectopia lentis; OR aortic root dilation/dissection AND FBN1 mutation; OR aortic root dilation/dissection AND systemic score ≥7; OR ectopia lentis AND FBN1 mutation known to cause aortic disease. The systemic score includes: wrist AND thumb sign (3), wrist OR thumb sign (1), pectus carinatum (2), pectus excavatum or chest asymmetry (1), hindfoot deformity (2), plain flat foot (1), pneumothorax (2), dural ectasia (2), protrusio acetabuli (2), reduced upper/lower segment ratio AND increased arm span/height (1), scoliosis or thoracolumbar kyphosis (1), reduced elbow extension (1), facial features 3/5 (dolichocephaly, enophthalmos, downslanting palpebral fissures, malar hypoplasia, retrognathia) (1), skin striae (1), myopia >3 diopters (1), mitral valve prolapse (1).
Management:
- Cardiovascular: beta-blockers (atenolol) or ARBs (losartan) to slow aortic root dilation; annual echocardiography; prophylactic aortic root replacement when diameter ≥5.0 cm (or ≥4.5 cm with risk factors: family history of dissection, rapid growth >0.5 cm/year, significant AR)
- Ophthalmologic: annual slit-lamp exam; surgical lens repositioning if needed
- Musculoskeletal: scoliosis monitoring, orthotics
- Activity restrictions: avoid contact sports, isometric exercise, competitive athletics
- Pregnancy: high risk of aortic dissection (especially if root >4.0 cm); requires multidisciplinary management
Ehlers-Danlos Syndromes (EDS)
A group of heritable connective tissue disorders characterized by joint hypermobility, skin hyperextensibility, and tissue fragility. The 2017 International Classification recognizes 13 subtypes.
| Type | Inheritance | Gene(s) | Key Features |
|---|---|---|---|
| Classical (cEDS) | AD | COL5A1, COL5A2 | Marked skin hyperextensibility, atrophic scarring, generalized joint hypermobility |
| Hypermobile (hEDS) | AD | Unknown | Most common type; generalized joint hypermobility, chronic pain, fatigue; no molecular test available; diagnosed clinically using 2017 criteria (Beighton score ≥5 adults, ≥6 children + systemic features) |
| Vascular (vEDS) | AD | COL3A1 | Most dangerous type; thin translucent skin, easy bruising, characteristic facies (thin lips/nose, large eyes), arterial/intestinal/uterine rupture; median survival ~50 years; avoid invasive vascular procedures; celiprolol may reduce vascular events |
| Kyphoscoliotic (kEDS) | AR | PLOD1, FKBP14 | Severe hypotonia at birth, progressive kyphoscoliosis, ocular fragility (globe rupture) |
| Arthrochalasia (aEDS) | AD | COL1A1, COL1A2 | Bilateral congenital hip dislocation, severe joint hypermobility |
| Dermatosparaxis (dEDS) | AR | ADAMTS2 | Extreme skin fragility, redundant skin, severe bruising |
Vascular EDS (vEDS): Arterial dissection or rupture, bowel perforation, and uterine rupture during pregnancy are life-threatening complications. Suspect in any young patient with spontaneous arterial dissection or bowel perforation without obvious cause. Avoid arteriography and elective surgery when possible. COL3A1 testing is essential when suspected.
10 Neurocutaneous Syndromes (NF, TSC)
Neurofibromatosis Type 1 (NF1)
Autosomal dominant, caused by mutations in the NF1 gene (17q11.2) encoding neurofibromin, a RAS-GAP (tumor suppressor). Prevalence ~1 in 3,000. ~50% de novo. Complete penetrance by age 5 but highly variable expressivity.
NIH Diagnostic Criteria (revised 2021) — diagnosis requires ≥2 of:
- ≥6 café-au-lait macules (>5 mm prepubertal, >15 mm postpubertal)
- ≥2 neurofibromas of any type OR 1 plexiform neurofibroma
- Axillary or inguinal freckling (Crowe sign)
- Optic pathway glioma
- ≥2 Lisch nodules (iris hamartomas) or ≥2 choroidal abnormalities
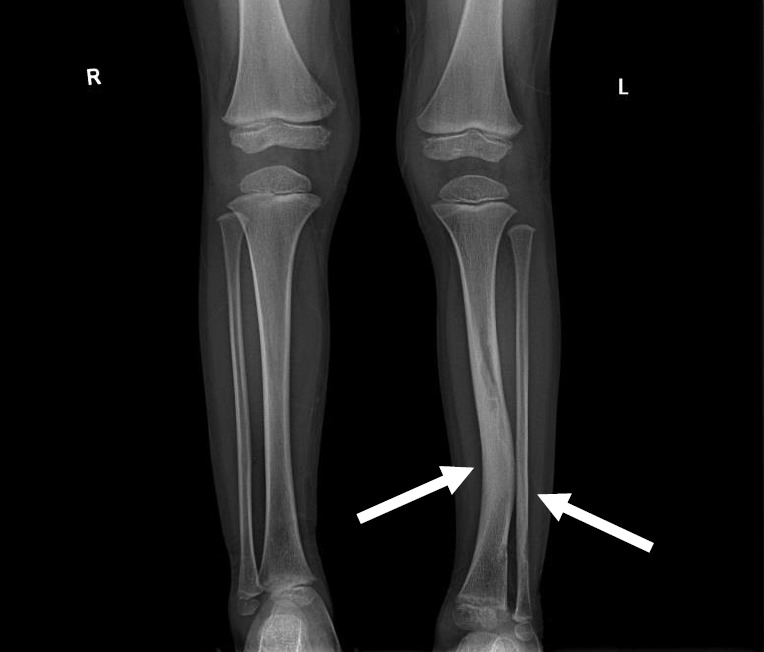
- Distinctive osseous lesion (sphenoid dysplasia, tibial pseudarthrosis, long bone bowing)
- First-degree relative with NF1 by above criteria
- Heterozygous pathogenic NF1 variant
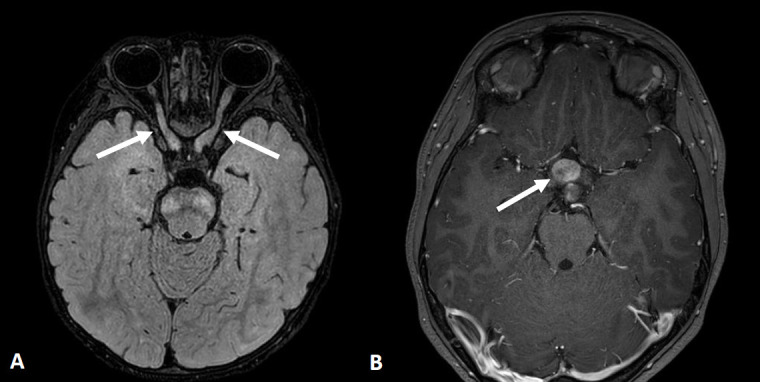
Complications: plexiform neurofibromas (can undergo malignant transformation to MPNST in ~8–13%), learning disabilities (~50–75%), scoliosis, hypertension (renal artery stenosis, pheochromocytoma), brain tumors. Surveillance: annual comprehensive exam, ophthalmologic screening, blood pressure monitoring, MRI as clinically indicated. Selumetinib (MEK inhibitor) approved for symptomatic, inoperable plexiform neurofibromas in children ≥2 years.
Neurofibromatosis Type 2 (NF2)
Autosomal dominant, caused by mutations in NF2 (merlin/schwannomin) on 22q12.2. Much rarer than NF1 (~1 in 25,000). Hallmark: bilateral vestibular schwannomas (presenting with hearing loss, tinnitus, balance problems, usually by age 20–30). Associated with meningiomas, ependymomas, and posterior subcapsular cataracts. Management: surgical resection, stereotactic radiosurgery, bevacizumab for progressive schwannomas; cochlear/brainstem implants for hearing.
Tuberous Sclerosis Complex (TSC)
Autosomal dominant, caused by mutations in TSC1 (hamartin, 9q34) or TSC2 (tuberin, 16p13.3). These form a complex that inhibits mTOR signaling; loss leads to uncontrolled cell growth and hamartoma formation in multiple organs. Prevalence ~1 in 6,000–10,000. ~2/3 de novo.
2021 Updated Diagnostic Criteria — Definite TSC: 2 major criteria OR 1 major + ≥2 minor OR pathogenic TSC1/TSC2 variant. Possible TSC: 1 major OR ≥2 minor.
Major criteria: (1) Hypomelanotic macules (≥3, at least 5 mm — "ash-leaf spots," best seen with Wood lamp); (2) Angiofibromas (≥3) or fibrous cephalic plaque; (3) Ungual fibromas (≥2); (4) Shagreen patch; (5) Multiple retinal hamartomas; (6) Cortical dysplasias (including tubers and radial migration lines); (7) Subependymal nodules (SENs); (8) Subependymal giant cell astrocytoma (SEGA); (9) Cardiac rhabdomyoma; (10) Lymphangioleiomyomatosis (LAM); (11) Renal angiomyolipomas (≥2).
Minor criteria: (1) "Confetti" skin lesions; (2) Dental enamel pits (>3); (3) Intraoral fibromas (≥2); (4) Retinal achromic patch; (5) Multiple renal cysts; (6) Nonrenal hamartomas; (7) Sclerotic bone lesions.
Management: mTOR inhibitors (everolimus) for SEGA, renal angiomyolipomas, and LAM; antiepileptic medications (vigabatrin is first-line for infantile spasms in TSC); regular surveillance of brain (MRI), kidneys (MRI or ultrasound), lungs (CT for LAM in women), heart (echo), skin, eyes, and neurodevelopment.
11 Trinucleotide Repeat & Other AD Disorders
Huntington Disease
Autosomal dominant neurodegenerative disorder caused by CAG trinucleotide repeat expansion in HTT gene (4p16.3). The expanded polyglutamine tract causes toxic gain of function. Prevalence ~5–10 per 100,000 in European populations.
| CAG Repeat Length | Classification | Clinical Significance |
|---|---|---|
| ≤26 | Normal | No disease risk |
| 27–35 | Intermediate | No symptoms but unstable; may expand in next generation (especially paternal transmission) |
| 36–39 | Reduced penetrance | May or may not develop disease |
| ≥40 | Full penetrance | Disease will manifest if individual lives long enough |
| ≥60 | Juvenile onset | Onset before age 20; rigid form predominates |
Clinical features: onset typically 30–50 years with progressive chorea (involuntary, irregular, non-patterned movements), psychiatric symptoms (depression, irritability, personality changes — often precede motor symptoms), and cognitive decline progressing to dementia. Juvenile form (Westphal variant) presents with rigidity, bradykinesia, and seizures rather than chorea. Anticipation is marked, especially with paternal transmission (spermatogenesis predisposes to further expansion). Death typically 15–20 years after onset.
Achondroplasia
Most common skeletal dysplasia causing dwarfism (~1 in 15,000–40,000). Autosomal dominant, caused by a gain-of-function mutation in FGFR3 (fibroblast growth factor receptor 3) on 4p16.3. ~80% are de novo mutations; virtually all caused by the same Gly380Arg variant. Advanced paternal age is a risk factor. Features: rhizomelic limb shortening, macrocephaly with frontal bossing, midface hypoplasia, trident hands, lumbar lordosis, average adult height ~131 cm (males) and ~124 cm (females). Complications: foramen magnum stenosis (infants), spinal stenosis (adults), recurrent otitis media, obstructive sleep apnea. Vosoritide (C-type natriuretic peptide analog) is FDA-approved for children ≥5 years to increase growth velocity.
Familial Hypercholesterolemia (FH)
Autosomal dominant (codominant). Mutations in LDLR (most common, ~85–90%), APOB (~5–10%), or PCSK9 (gain of function, ~1%). Heterozygous FH prevalence ~1 in 250; homozygous ~1 in 300,000. Heterozygous FH: LDL-C 190–400 mg/dL, tendon xanthomas (especially Achilles), corneal arcus, premature ASCVD (MI by age 40–50 in males). Homozygous FH: LDL-C >500 mg/dL, cutaneous xanthomas in childhood, ASCVD in childhood/adolescence, aortic valve disease. Dutch Lipid Clinic Network criteria used for clinical diagnosis. Management: high-intensity statins, ezetimibe, PCSK9 inhibitors (evolocumab, alirocumab), LDL apheresis for homozygous FH.
Myotonic Dystrophy Type 1 (DM1)
Autosomal dominant, caused by CTG trinucleotide repeat expansion in DMPK gene on 19q13.3. Most common adult muscular dystrophy (~1 in 8,000). Normal: 5–34 repeats; premutation: 35–49; mild: 50–150; classic: 100–1,000; congenital: >1,000. Clinical features: myotonia (delayed relaxation after muscle contraction), progressive muscle weakness and wasting (distal > proximal; facial, neck, and hand muscles first), cataracts (posterior subcapsular, "Christmas tree" pattern), cardiac conduction defects (first-degree AV block, bundle branch block — sudden death risk), frontal balding, testicular atrophy, insulin resistance. Congenital DM1 (maternally transmitted with massive expansions) presents with severe neonatal hypotonia, respiratory failure, and intellectual disability. Marked anticipation, especially with maternal transmission.
Hereditary Spherocytosis
Most commonly autosomal dominant (~75%). Mutations in red cell membrane/cytoskeletal proteins: ankyrin (ANK1, most common), band 3 (SLC4A1), spectrin (α or β), protein 4.2. Spherical, osmotically fragile RBCs are trapped and destroyed in the spleen. Features: hemolytic anemia (variable severity), jaundice, splenomegaly, pigmented gallstones. Diagnosis: positive osmotic fragility test or eosin-5-maleimide (EMA) binding test. Management: folate supplementation, splenectomy for severe cases (ideally after age 6, with prior vaccination against encapsulated organisms).
Polycystic Kidney Disease (ADPKD)
Autosomal dominant, caused by mutations in PKD1 (16p13.3, ~85%, more severe) or PKD2 (4q22.1, ~15%, milder course). Most common hereditary kidney disease (~1 in 400–1,000). Progressive bilateral renal cysts leading to ESRD (median age ~55 for PKD1, ~75 for PKD2). Extrarenal manifestations: hepatic cysts (~80%), intracranial aneurysms (~5–10% — screen with MRA if family history of rupture or SAH), mitral valve prolapse, diverticulosis. Tolvaptan (V2 receptor antagonist) slows cyst growth and GFR decline in rapidly progressive disease.
12 Cystic Fibrosis
Autosomal recessive disorder caused by mutations in the CFTR gene (7q31.2) encoding the cystic fibrosis transmembrane conductance regulator, a chloride/bicarbonate channel on epithelial cell surfaces. Most common life-shortening AR disorder in Caucasians (~1 in 2,500–3,500 births; carrier frequency ~1 in 25). Over 2,000 CFTR variants identified; F508del (p.Phe508del) accounts for ~70% of pathogenic alleles worldwide.
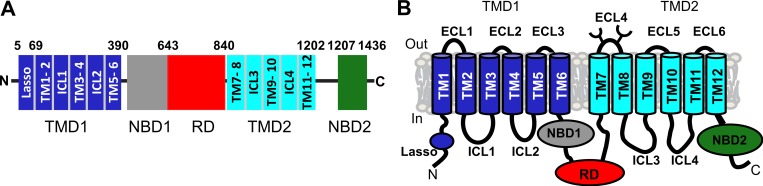
CFTR Mutation Classes
| Class | Defect | Example | Severity |
|---|---|---|---|
| I | No protein synthesis (nonsense, frameshift) | G542X, W1282X | Severe |
| II | Protein misfolding/trafficking defect | F508del | Severe |
| III | Gating defect (channel does not open) | G551D | Severe |
| IV | Reduced conductance (channel opens but Cl− flow reduced) | R117H | Mild |
| V | Reduced protein quantity (splicing defects) | 3849+10kbC→T | Mild |
| VI | Decreased surface stability | rF508del (rescued) | Variable |
Clinical Features
- Pulmonary: thick, dehydrated airway mucus → chronic bacterial infections (Staphylococcus aureus early, Pseudomonas aeruginosa later, Burkholderia cepacia complex — poor prognosis), bronchiectasis, progressive obstructive lung disease (leading cause of death)
- GI/Pancreatic: pancreatic insufficiency (~85%, presents with steatorrhea, fat-soluble vitamin deficiency — A, D, E, K), meconium ileus (~15% of neonates — virtually pathognomonic for CF), distal intestinal obstruction syndrome (DIOS), CF-related diabetes (CFRD, ~20% of adolescents, ~50% of adults), biliary cirrhosis
- Reproductive: males — congenital bilateral absence of vas deferens (CBAVD) causing infertility (~98%); females — reduced fertility from thick cervical mucus
- Other: nasal polyps, sinusitis, digital clubbing, CF-related bone disease
Diagnosis
Newborn screening: immunoreactive trypsinogen (IRT) on dried blood spot, followed by CFTR mutation analysis and/or repeat IRT. Confirmed by sweat chloride test (gold standard): ≥60 mmol/L diagnostic, 30–59 mmol/L intermediate, ≤29 mmol/L normal. CFTR genotyping identifies specific mutations for prognostic and therapeutic purposes.

CFTR Modulator Therapy
| Drug | Type | Target Mutations | Details |
|---|---|---|---|
| Ivacaftor (Kalydeco) | Potentiator (opens channel) | G551D and other gating mutations (Class III) | Monotherapy for gating mutations; dramatic improvement in sweat Cl−, FEV1, weight; age ≥4 months |
| Lumacaftor/ivacaftor (Orkambi) | Corrector + potentiator | F508del homozygous | Modest FEV1 improvement (~2.6–4%); age ≥2 years |
| Tezacaftor/ivacaftor (Symdeko) | Corrector + potentiator | F508del homozygous or F508del + residual function | Better tolerated than Orkambi; age ≥6 years |
| Elexacaftor/tezacaftor/ivacaftor (Trikafta) | Triple combination (2 correctors + potentiator) | At least one F508del allele (~90% of CF patients eligible) | Transformative: FEV1 improvement ~14%, sweat Cl− reduction ~40 mmol/L, reduced pulmonary exacerbations by ~60%; age ≥2 years. Has changed the natural history of CF |
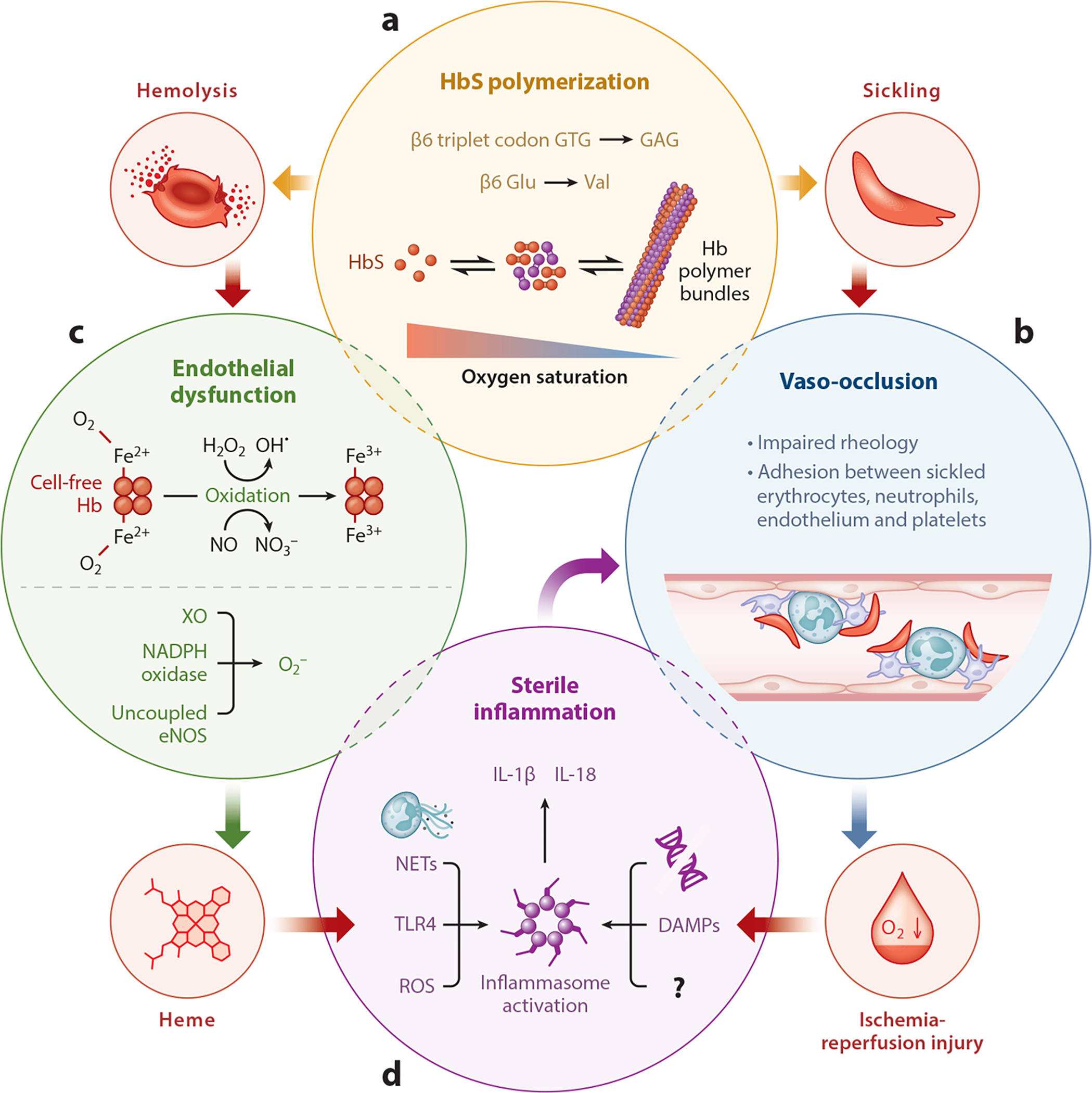
13 Sickle Cell Disease & Hemoglobinopathies
Sickle cell disease (SCD) is caused by a missense mutation in the HBB gene (11p15.4): Glu6Val (GAG → GTG) producing hemoglobin S (HbS). Autosomal recessive. Most common monogenic disorder worldwide; carrier frequency ~8–10% in African Americans.
SCD Genotypes
| Genotype | Designation | Severity | Hb Electrophoresis |
|---|---|---|---|
| HbSS | Sickle cell anemia | Most severe | HbS ~80–90%, HbF 2–20%, HbA2 2–4%, HbA absent |
| HbSC | Hemoglobin SC disease | Moderate | HbS ~50%, HbC ~50%; higher risk of retinopathy, AVN |
| HbS/β0-thal | Sickle-beta-zero thalassemia | Severe (like SS) | HbS ~80–90%, HbF 2–15%, HbA absent, elevated HbA2 |
| HbS/β+-thal | Sickle-beta-plus thalassemia | Mild to moderate | HbS 60–80%, HbA 10–30%, elevated HbA2 |
Complications
- Vaso-occlusive crisis (VOC): most common reason for hospitalization; dactylitis (hand-foot syndrome) often first manifestation in infants <2 years
- Acute chest syndrome: new infiltrate + respiratory symptoms ± fever; leading cause of death in adults; triggered by infection, fat embolism from bone marrow, or hypoventilation
- Stroke: ~11% by age 20 without prevention; transcranial Doppler (TCD) screening annually ages 2–16; chronic transfusion therapy if TCD velocity ≥200 cm/s
- Splenic sequestration: acute splenic enlargement with hemoglobin drop ≥2 g/dL; can be fatal; functional asplenia by age 5 in HbSS — risk of overwhelming sepsis from encapsulated organisms
- Aplastic crisis: parvovirus B19 infection → transient red cell aplasia → acute severe anemia
- Chronic organ damage: avascular necrosis (especially femoral head), proliferative retinopathy, chronic kidney disease, pulmonary hypertension, leg ulcers, priapism, cholelithiasis
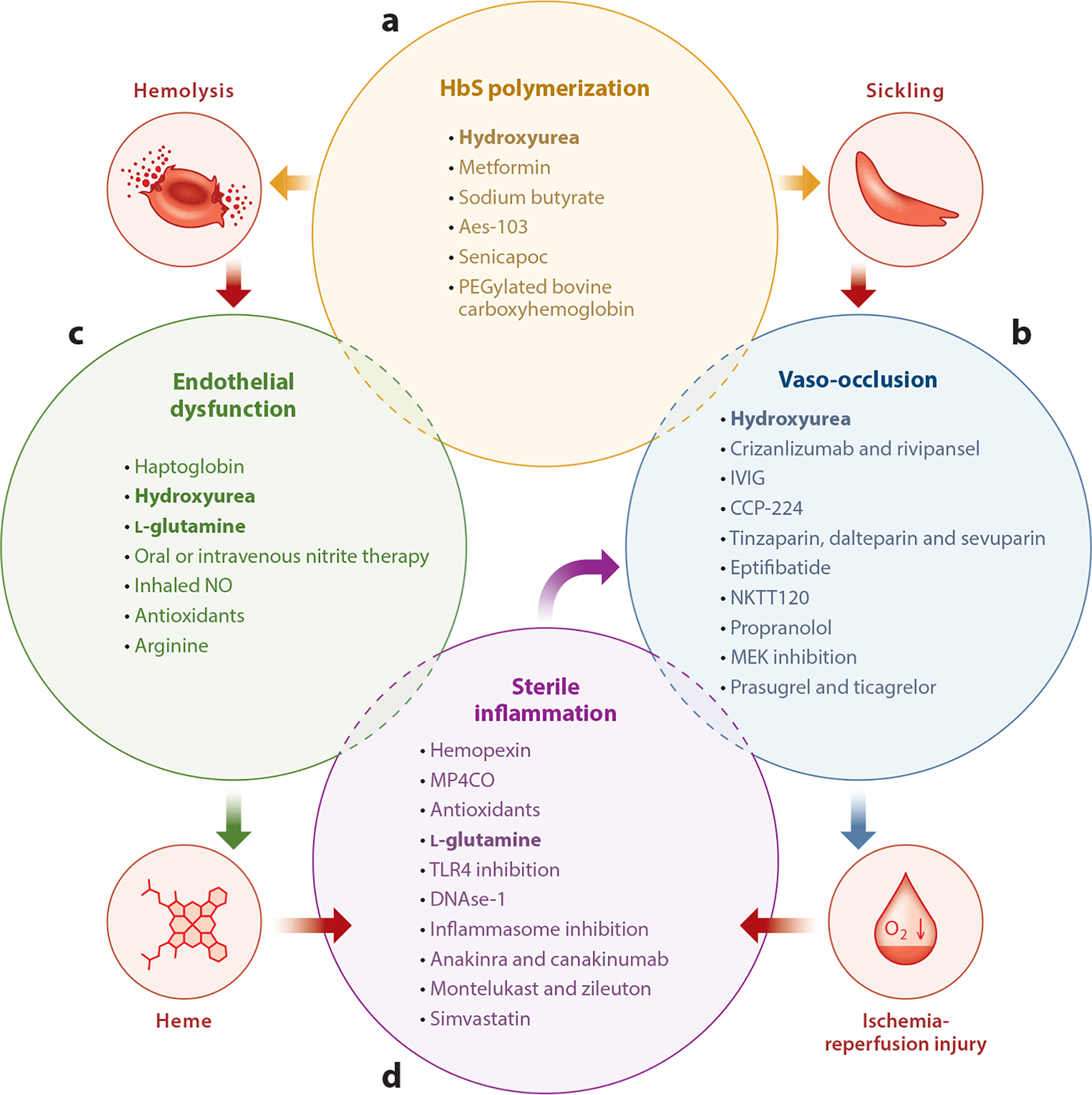
Management
Hydroxyurea: increases HbF production; reduces VOC by ~50%, reduces ACS, reduces need for transfusion; recommended for all patients ≥9 months with HbSS or HbS/β0-thal. L-glutamine (Endari): reduces oxidative stress in sickled RBCs; adjunctive to hydroxyurea. Crizanlizumab: anti-P-selectin antibody, reduces VOC. Voxelotor: HbS polymerization inhibitor, improves Hb. Chronic transfusion: target HbS <30% for primary/secondary stroke prevention. Hematopoietic stem cell transplant: only curative option (matched sibling donor); >90% cure rate. Gene therapy: lovotibeglogene autotemcel (Lyfgenia, lentiviral) and exagamglogene autotemcel (Casgevy, CRISPR-based — first FDA-approved CRISPR therapy) approved for severe SCD in patients ≥12 years.
14 PKU, SMA & Other AR Disorders
Phenylketonuria (PKU)
Caused by mutations in PAH (phenylalanine hydroxylase) gene on 12q23.2. Inability to convert phenylalanine (Phe) to tyrosine → Phe accumulation causes intellectual disability if untreated. Newborn screening detects elevated Phe on dried blood spot (all 50 US states since 1960s). Classification: classic PKU (Phe >1,200 μmol/L untreated), moderate PKU (600–1,200), mild PKU (360–600), mild HPA (<360, often does not require treatment). Treatment: Phe-restricted diet (lifelong), supplemented with medical formula; target blood Phe 120–360 μmol/L. Sapropterin (BH4, Kuvan): cofactor for PAH, effective in ~25–50% of patients (mostly milder forms). Pegvaliase (Palynziq): enzyme substitution for adults with uncontrolled PKU. Maternal PKU: uncontrolled maternal Phe during pregnancy causes microcephaly, congenital heart defects, and intellectual disability in the fetus (regardless of fetal genotype) — strict dietary control before and throughout pregnancy is essential.
Spinal Muscular Atrophy (SMA)
Caused by homozygous deletion or mutation of SMN1 gene on 5q13. SMN2 copy number modifies severity (more copies = milder phenotype). AR; carrier frequency ~1 in 50. Prevalence ~1 in 10,000.
| Type | Onset | Motor Milestone | SMN2 Copies | Natural History |
|---|---|---|---|---|
| Type 0 | Prenatal | None | 1 | Severe arthrogryposis, respiratory failure at birth, death within weeks |
| Type I (Werdnig-Hoffmann) | <6 months | Never sit | 1–2 | Most common (~60%); severe hypotonia, tongue fasciculations, bell-shaped chest, respiratory failure; death by age 2 without treatment |
| Type II (Dubowitz) | 6–18 months | Sit independently, never walk | 3 | Progressive scoliosis, respiratory insufficiency; survival into adulthood with supportive care |
| Type III (Kugelberg-Welander) | >18 months | Walk independently (may lose) | 3–4 | Proximal weakness, Gowers sign; normal lifespan |
| Type IV | Adulthood (>21 years) | Walk independently | 4–8 | Mild proximal weakness; normal lifespan |
Therapies: Nusinersen (Spinraza): antisense oligonucleotide that modifies SMN2 splicing to increase functional SMN protein; intrathecal injection every 4 months after loading doses. Onasemnogene abeparvovec (Zolgensma): AAV9-based gene therapy delivering functional SMN1 gene; single IV infusion; approved for age <2 years; one of the most expensive drugs ever (>$2M). Risdiplam (Evrysdi): oral SMN2 splicing modifier; approved for age ≥2 months. Pre-symptomatic treatment (identified by newborn screening) has shown the best outcomes, with many treated infants achieving near-normal motor development.
Other Key AR Disorders
Tay-Sachs disease: HEXA gene mutations → deficiency of hexosaminidase A → GM2 ganglioside accumulation in neurons. Infantile form: normal development until ~6 months, then progressive neurodegeneration, cherry-red spot on macula, seizures, macrocephaly, death by age 3–5. High carrier frequency in Ashkenazi Jewish (~1 in 30), French-Canadian, and Cajun populations. Carrier screening recommended.
Gaucher disease: GBA gene → deficiency of glucocerebrosidase → glucocerebroside accumulation in macrophages ("Gaucher cells"). Type I (non-neuronopathic, most common): hepatosplenomegaly, bone disease (Erlenmeyer flask deformity, avascular necrosis), cytopenias; does NOT affect the brain; treated with enzyme replacement therapy (imiglucerase, velaglucerase alfa) or substrate reduction therapy (eliglustat, miglustat). Type II (acute neuronopathic): severe CNS involvement, death by age 2. Type III (chronic neuronopathic): intermediate CNS involvement, variable survival. GBA heterozygous carriers have 5–8× increased risk of Parkinson disease.
Wilson disease: ATP7B gene → defective copper transport → copper accumulation in liver (hepatitis, cirrhosis), brain (movement disorders, psychiatric symptoms), and cornea (Kayser-Fleischer rings). Low serum ceruloplasmin (<20 mg/dL), elevated 24-hour urine copper (>100 μg/day), elevated hepatic copper (>250 μg/g dry weight). Treatment: chelation with D-penicillamine or trientine; zinc (blocks intestinal absorption); liver transplant for fulminant hepatic failure.
Hereditary hemochromatosis: HFE gene; C282Y homozygosity accounts for ~80–90% of cases in European populations. Iron overload → liver cirrhosis (increased HCC risk), diabetes ("bronze diabetes"), cardiomyopathy, arthropathy (2nd and 3rd MCP joints), hypogonadism, skin hyperpigmentation. Low penetrance (~10–25% of C282Y homozygotes develop clinical disease, higher in males). Diagnosis: elevated transferrin saturation (>45%), elevated ferritin; HFE genotyping. Treatment: phlebotomy (target ferritin 50–100 μg/L).
15 Duchenne & Becker Muscular Dystrophy
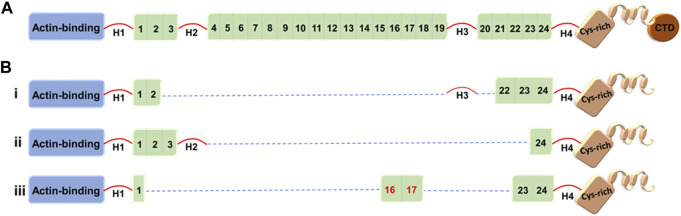
X-linked recessive disorders caused by mutations in the DMD gene (Xp21.2), the largest known human gene (2.4 Mb, 79 exons), encoding dystrophin, a critical structural protein linking the cytoskeleton to the extracellular matrix in muscle fibers.
Duchenne Muscular Dystrophy (DMD)
Incidence ~1 in 3,500–5,000 live male births. Caused by out-of-frame mutations (usually large deletions) that abolish dystrophin production. Clinical features: normal early development; progressive proximal muscle weakness beginning age 2–5; Gowers sign (using hands to "walk up" the legs when rising from floor); calf pseudohypertrophy (replacement of muscle with fat/fibrotic tissue); loss of ambulation by age 12; cardiomyopathy (dilated); respiratory failure; death in 20s–30s without ventilatory support. CK massively elevated (10,000–50,000+ U/L). Diagnosis confirmed by genetic testing (multiplex ligation-dependent probe amplification — MLPA, or sequencing).
Becker Muscular Dystrophy (BMD)
Caused by in-frame mutations that produce a partially functional (truncated or partially functional) dystrophin protein. Later onset (typically age 5–15), slower progression, ambulation often maintained into 20s–40s, cardiomyopathy can be the predominant feature. CK elevated but typically lower than DMD.
Out-of-frame deletions (disrupt the reading frame) → no functional dystrophin → Duchenne (severe). In-frame deletions (preserve the reading frame) → partially functional dystrophin → Becker (milder). This rule holds in ~90% of cases and is the basis for exon-skipping therapy strategies.
Management:
- Corticosteroids: deflazacort or prednisone (standard of care) — prolongs ambulation by ~2–3 years, delays scoliosis and cardiomyopathy
- Cardiac: ACE inhibitors or ARBs starting at diagnosis or by age 10 (before cardiomyopathy onset); beta-blockers if LV dysfunction develops
- Respiratory: regular PFTs; nocturnal non-invasive ventilation when FVC <50% predicted
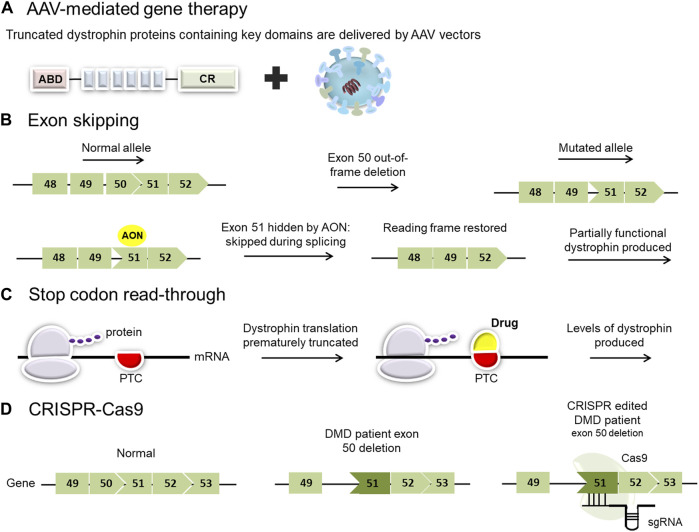
- Exon-skipping therapies: antisense oligonucleotides that restore the reading frame for specific deletions — eteplirsen (exon 51, ~13% of patients), golodirsen and viltolarsen (exon 53, ~8%), casimersen (exon 45, ~8%); approved under accelerated approval, modest dystrophin restoration
- Gene therapy: delandistrogene moxeparvovec (Elevidys, AAV-based micro-dystrophin); approved for ages 4–5 years
16 Hemophilia, G6PD & Other X-Linked Disorders
Hemophilia A & B
X-linked recessive bleeding disorders. Hemophilia A: deficiency of factor VIII (F8 gene, Xq28); ~1 in 5,000 males; ~50% due to intron 22 inversion. Hemophilia B (Christmas disease): deficiency of factor IX (F9 gene, Xq27.1); ~1 in 25,000 males.
| Severity | Factor Level | Bleeding Pattern |
|---|---|---|
| Severe | <1% (<0.01 IU/mL) | Spontaneous joint/muscle bleeds (hemarthroses); frequent, starting in infancy |
| Moderate | 1–5% | Bleeding with minor trauma or surgery; occasional spontaneous bleeds |
| Mild | 5–40% | Bleeding with significant trauma or surgery; may not be diagnosed until adulthood |
Management: factor replacement (recombinant preferred); prophylaxis for severe disease (regular infusions to maintain trough >1%). Extended half-life factor products reduce infusion frequency. Emicizumab (Hemlibra): bispecific antibody bridging FIXa and FX (mimics FVIII function); subcutaneous, every 1–4 weeks; revolutionary for hemophilia A prophylaxis, including patients with inhibitors. Fitusiran: anti-antithrombin siRNA for hemophilia A or B. Gene therapy: valoctocogene roxaparvovec (Roctavian, AAV5-FVIII) for hemophilia A; etranacogene dezaparvovec (Hemgenix, AAV5-FIX Padua) for hemophilia B.
G6PD Deficiency
X-linked recessive. Most common enzymopathy worldwide (~400 million affected). Glucose-6-phosphate dehydrogenase is essential for generating NADPH via the pentose phosphate pathway, which protects red cells from oxidative damage. Carrier advantage: partial protection against Plasmodium falciparum malaria.
Triggers of hemolytic crisis: infections (most common trigger), fava beans (favism), medications (primaquine, dapsone, sulfonamides, nitrofurantoin, rasburicase, methylene blue). Hemolysis pattern: acute intravascular hemolysis 24–72 hours after exposure; Heinz bodies (denatured Hb inclusions) and bite cells on peripheral smear; self-limited as older, more vulnerable RBCs are destroyed and replaced by younger cells with higher G6PD activity. G6PD assay may be falsely normal during acute hemolysis (test after recovery). Management: avoid triggers; supportive care; transfusion if severe anemia.
Fabry Disease
X-linked (affects males severely; carrier females can have mild-moderate disease due to skewed X-inactivation). GLA gene → deficiency of α-galactosidase A → accumulation of globotriaosylceramide (Gb3) in vascular endothelium, kidneys, heart, and nervous system. Classic presentation in males: acroparesthesias (burning pain in hands/feet, childhood onset), angiokeratomas (skin), corneal verticillata (whorl-like opacities), hypohidrosis, progressive renal failure, cardiomyopathy (LVH), stroke. Treatment: enzyme replacement therapy (agalsidase alfa or beta) or oral chaperone therapy (migalastat for amenable GLA mutations).
Hunter Syndrome (MPS II)
X-linked recessive. IDS gene → deficiency of iduronate-2-sulfatase → accumulation of heparan sulfate and dermatan sulfate. Features: coarse facies, hepatosplenomegaly, dysostosis multiplex, joint stiffness, cardiac valve disease, hearing loss. Neuronopathic (severe) form: progressive intellectual disability, death in teens. Non-neuronopathic (attenuated) form: normal intelligence, survival to adulthood. Treatment: enzyme replacement (idursulfase); HSCT considered for severe form if diagnosed early.
Lesch-Nyhan Syndrome
X-linked recessive. HPRT1 gene → deficiency of hypoxanthine-guanine phosphoribosyltransferase → massive overproduction of uric acid. Features: hyperuricemia/gout, nephrolithiasis, intellectual disability, and characteristic self-injurious behavior (lip and finger biting, head banging). Treatment: allopurinol for hyperuricemia (does not affect neurologic features); behavioral interventions.
17 Fragile X & Trinucleotide X-Linked Disorders
Fragile X Syndrome
Most common inherited cause of intellectual disability and most common monogenic cause of autism spectrum disorder. Caused by CGG trinucleotide repeat expansion in the 5′ UTR of the FMR1 gene (Xq27.3), leading to promoter hypermethylation and gene silencing (→ absence of FMRP, a protein essential for synaptic plasticity).
| CGG Repeat Range | Category | Clinical Significance |
|---|---|---|
| 5–44 | Normal | No disease; stable transmission |
| 45–54 | Intermediate (gray zone) | No disease; may expand to premutation in next generation |
| 55–200 | Premutation | No fragile X syndrome but: females — premature ovarian insufficiency (FXPOI, ~20%); both sexes — fragile X-associated tremor/ataxia syndrome (FXTAS, late onset, especially males >50 years); maternal premutations can expand to full mutation in offspring (risk correlates with repeat size; ≥90 repeats → nearly 100% expansion risk) |
| >200 | Full mutation | Fragile X syndrome: males — moderate intellectual disability (IQ 35–70), characteristic facies (long face, prominent ears, prominent jaw), macroorchidism (postpubertal), ADHD, anxiety, ASD (~30%); carrier females — ~50% have mild intellectual impairment due to X-inactivation patterns |
Rett Syndrome
X-linked dominant disorder caused by mutations in MECP2 (Xq28). Almost exclusively affects females (~1 in 10,000–15,000 female births); usually lethal in males. ~99% de novo. Clinical course: apparently normal development until 6–18 months → developmental regression (loss of purposeful hand use, loss of spoken language) → characteristic stereotypic hand movements (wringing, clapping, mouthing) → acquired microcephaly, seizures (~80%), scoliosis, autonomic dysfunction (breathing irregularities, cardiac QT prolongation), gait apraxia. Classified by classic Rett (4 stages) and atypical variants. Management is supportive; trofinetide (Daybue) is the first FDA-approved treatment for Rett syndrome.
18 Mitochondrial Disorders
Mitochondrial disorders can be caused by mutations in mitochondrial DNA (mtDNA) or nuclear DNA genes encoding mitochondrial proteins. mtDNA-encoded disorders follow maternal inheritance (mother transmits to all children; father cannot transmit). The clinical phenotype depends on the mutation, the proportion of mutant mtDNA (heteroplasmy), and the energy demands of affected tissues (brain, muscle, heart, and retina are most vulnerable).
Key Mitochondrial Disorders
| Disorder | Mutation | Key Features |
|---|---|---|
| MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis, Stroke-like episodes) | m.3243A>G (MT-TL1, tRNA-Leu) in ~80% | Stroke-like episodes (often occipital, not following vascular territories), seizures, migraine-like headaches, lactic acidosis, short stature, diabetes mellitus, sensorineural hearing loss, ragged red fibers on muscle biopsy |
| MERRF (Myoclonic Epilepsy with Ragged Red Fibers) | m.8344A>G (MT-TK, tRNA-Lys) in ~80% | Myoclonus, epilepsy, ataxia, ragged red fibers, lipomas, hearing loss |
| LHON (Leber Hereditary Optic Neuropathy) | m.11778G>A (MT-ND4) ~70%; m.14484T>C (MT-ND6); m.3460G>A (MT-ND1) | Acute/subacute bilateral painless vision loss (central scotoma), males affected 4× more than females (incomplete penetrance, possibly estrogen-protective); onset typically ages 15–35; no ragged red fibers |
| Kearns-Sayre syndrome | Large mtDNA deletions (single, sporadic — not maternally inherited) | Onset <20 years; progressive external ophthalmoplegia (PEO), pigmentary retinopathy, cardiac conduction defects (may need pacemaker), cerebellar ataxia, elevated CSF protein |
| NARP (Neuropathy, Ataxia, Retinitis Pigmentosa) | m.8993T>G or T>C (MT-ATP6) | Sensory neuropathy, ataxia, retinitis pigmentosa; >90% mutant load → Leigh syndrome (fatal infantile necrotizing encephalopathy) |
| Pearson syndrome | Large mtDNA deletions | Sideroblastic anemia, pancreatic insufficiency in infancy; survivors often develop Kearns-Sayre |
Diagnostic approach: elevated lactate (blood and/or CSF), elevated lactate:pyruvate ratio (>20), brain MRI (stroke-like lesions, basal ganglia abnormalities in Leigh syndrome), muscle biopsy (ragged red fibers with modified Gomori trichrome, COX-negative fibers), mtDNA testing (blood or muscle). Genetic confirmation through mtDNA sequencing or targeted testing.
Treatment: largely supportive. Coenzyme Q10 (ubiquinone), riboflavin, L-carnitine, and alpha-lipoic acid are commonly used though evidence is limited. Idebenone has shown benefit for LHON vision recovery. Avoidance of mitochondrial toxins (valproate, aminoglycosides, statins at high doses). Cardiac pacing for conduction defects. Seizure management (avoid valproate in POLG mutations). Genetic counseling is complex due to heteroplasmy and the bottleneck effect in maternal transmission.
19 Amino Acid & Organic Acid Disorders
Amino Acid Disorders
| Disorder | Gene/Enzyme | Key Features | Treatment |
|---|---|---|---|
| Phenylketonuria (PKU) | PAH / phenylalanine hydroxylase | Intellectual disability, fair skin/hair (reduced melanin), musty odor, eczema, seizures (if untreated) | Low-Phe diet, sapropterin, pegvaliase |
| Maple syrup urine disease (MSUD) | BCKDHA/B/DBT / branched-chain α-ketoacid dehydrogenase | Sweet maple syrup odor, poor feeding, encephalopathy, metabolic acidosis; neonatal onset can be fatal | Dietary restriction of leucine, isoleucine, valine; liver transplant for severe forms |
| Homocystinuria | CBS / cystathionine β-synthase (most common) | Marfanoid habitus but osteoporosis, lens subluxation (inferonasal — vs. superotemporal in Marfan), intellectual disability, thromboembolism (leading cause of death) | Pyridoxine (B6) responsive in ~50%; methionine-restricted diet, betaine |
| Tyrosinemia type I | FAH / fumarylacetoacetate hydrolase | Hepatorenal disease, "cabbage-like" odor, rickets, hepatocellular carcinoma risk, porphyria-like crises | Nitisinone (NTBC) + low-tyrosine/phenylalanine diet; liver transplant if refractory |
| Alkaptonuria | HGD / homogentisic acid oxidase | Dark urine (on standing), ochronosis (blue-black pigmentation of cartilage), arthritis | Nitisinone; low-protein diet |
Organic Acidemias
| Disorder | Gene/Enzyme | Presentation | Diagnosis | Treatment |
|---|---|---|---|---|
| Propionic acidemia | PCCA/PCCB / propionyl-CoA carboxylase | Neonatal: metabolic acidosis, hyperammonemia, ketosis, pancytopenia; long-term: cardiomyopathy, movement disorders | Elevated propionylcarnitine (C3) on NBS; urine organic acids (3-hydroxypropionate, methylcitrate) | Protein-restricted diet (low isoleucine, valine, methionine, threonine); carnitine; metronidazole (reduces propionate-producing gut bacteria); liver transplant |
| Methylmalonic acidemia | MUT / methylmalonyl-CoA mutase (mut0 or mut−) | Similar to propionic; also renal disease; may respond to B12 (cobalamin) | Elevated C3; urine methylmalonic acid | B12-responsive forms: hydroxocobalamin; protein restriction; carnitine; liver ± kidney transplant |
| Isovaleric acidemia | IVD / isovaleryl-CoA dehydrogenase | "Sweaty feet" odor; metabolic acidosis, pancytopenia | Elevated isovalerylcarnitine (C5); isovalerylglycine in urine | Leucine-restricted diet; glycine and carnitine supplementation |
Acute metabolic crisis in organic acidemias: Triggered by catabolic stress (illness, fasting, surgery). Presents with metabolic acidosis (high anion gap), hyperammonemia, hypoglycemia, and encephalopathy. Management: (1) Stop protein intake; (2) Provide high glucose infusion (D10 at 1.5× maintenance, ± insulin for glucose >200) to reverse catabolism; (3) Carnitine IV; (4) Correct acidosis with bicarbonate if pH <7.1; (5) If hyperammonemia >500 μmol/L or rising, start continuous renal replacement therapy (CRRT). Consult metabolic specialist immediately.
20 Urea Cycle Defects
The urea cycle converts toxic ammonia (from protein catabolism) to urea for renal excretion. Six enzymes are involved; deficiency of any leads to hyperammonemia. Overall incidence ~1 in 30,000. OTC deficiency (ornithine transcarbamylase, X-linked) is the most common (~1 in 14,000); all others are autosomal recessive.
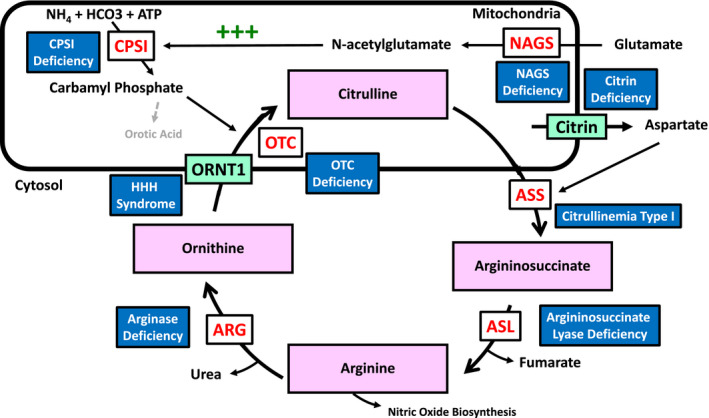
Urea Cycle Enzymes & Disorders
| Enzyme | Gene | Inheritance | Distinguishing Features |
|---|---|---|---|
| N-acetylglutamate synthase (NAGS) | NAGS | AR | Rarest; responds to carglumic acid (N-carbamylglutamate) |
| Carbamoyl phosphate synthetase I (CPS1) | CPS1 | AR | Low citrulline, low arginine |
| Ornithine transcarbamylase (OTC) | OTC | X-linked | Most common; elevated orotic acid in urine (distinguishes from CPS1); carrier females may have mild symptoms, especially postpartum |
| Argininosuccinate synthetase (ASS1) | ASS1 | AR | Citrullinemia type I; massively elevated citrulline |
| Argininosuccinate lyase (ASL) | ASL | AR | Argininosuccinic aciduria; elevated argininosuccinic acid; trichorrhexis nodosa (brittle hair) |
| Arginase (ARG1) | ARG1 | AR | Hyperargininemia; progressive spasticity rather than acute hyperammonemia; elevated arginine |
Clinical Presentation
Neonatal severe form (usually complete enzyme deficiency): presents within 24–72 hours of life with poor feeding, lethargy progressing to coma, tachypnea (respiratory alkalosis from central hyperventilation), seizures, and cerebral edema. Ammonia levels often >1,000 μmol/L (normal <50). Without treatment, rapid progression to brain damage and death.
Late-onset/partial deficiency: triggered by catabolic stress (intercurrent illness, high protein intake, postpartum state, valproate use). Presents with episodic vomiting, confusion, ataxia, psychiatric symptoms, and headaches. May be misdiagnosed as Reye syndrome, psychiatric illness, or cyclic vomiting.
Hyperammonemia management protocol:
- Stop all protein intake
- High-calorie IV glucose (D10–D25 at 1.5× maintenance, ± intralipid) to reverse catabolism
- Nitrogen scavengers: sodium benzoate (conjugates glycine) + sodium phenylacetate/phenylbutyrate (conjugates glutamine) — IV Ammonul for acute crisis
- Arginine IV (except in arginase deficiency) — provides substrate for urea cycle and depletes accumulated intermediates
- Carglumic acid for NAGS deficiency
- Hemodialysis/CRRT: if ammonia >500 μmol/L or not responding rapidly to medical management; peritoneal dialysis too slow for ammonia
- Monitor: ammonia levels every 2–4 hours initially; goal to reduce to <200 μmol/L within hours
Brain damage correlates with peak ammonia level and duration. Neonatal ammonia >1,000 μmol/L for >24 hours is associated with severe neurologic outcomes. Ammonia >300 μmol/L at any age requires urgent treatment.
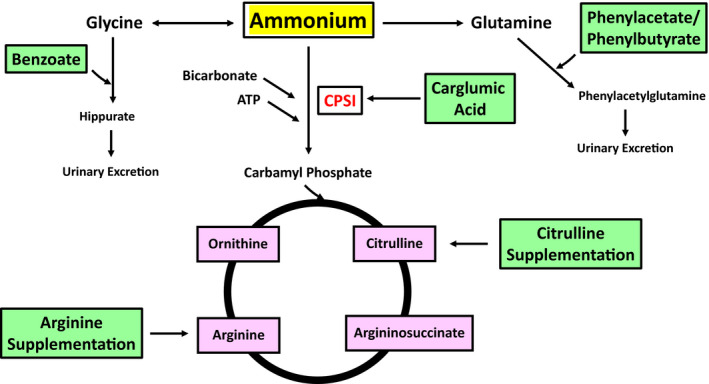
Long-term management: protein-restricted diet with essential amino acid supplementation, oral nitrogen scavengers (sodium phenylbutyrate or glycerol phenylbutyrate), arginine or citrulline supplementation, sick-day protocols (increase calories, reduce protein, have emergency letter). Liver transplant is curative for severe defects (corrects the enzymatic deficiency in the liver but does not reverse existing brain injury).
21 Fatty Acid Oxidation Disorders
Mitochondrial fatty acid oxidation (FAO) provides energy during fasting and prolonged exercise by converting fatty acids to acetyl-CoA for ketone body production. FAO disorders present with hypoketotic hypoglycemia (the hallmark — inability to generate ketones during fasting), hepatopathy, cardiomyopathy, and/or rhabdomyolysis.
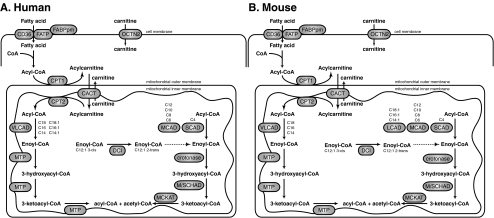
| Disorder | Enzyme | Gene | NBS Marker | Key Features |
|---|---|---|---|---|
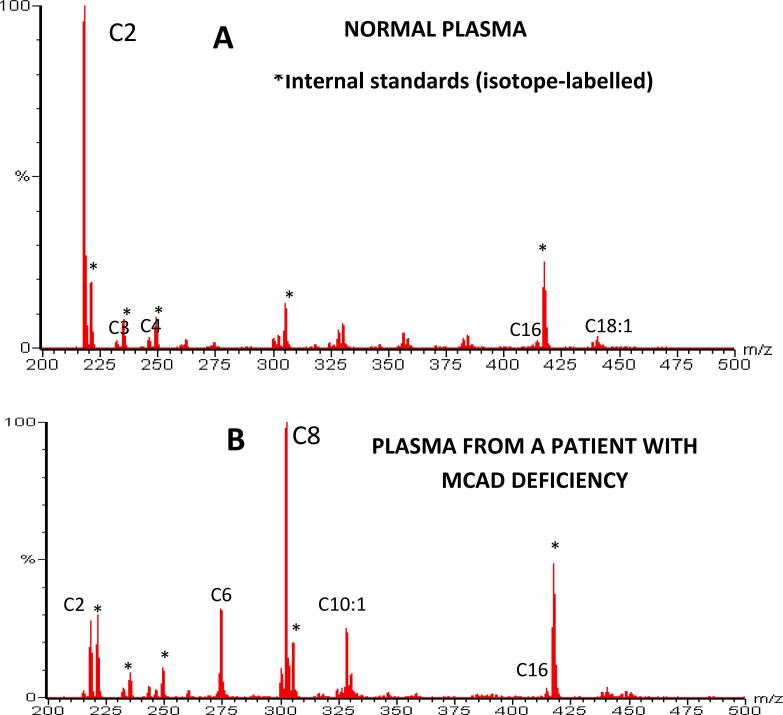
| MCADD (medium-chain acyl-CoA dehydrogenase deficiency) | MCAD | ACADM | Elevated C8 (octanoylcarnitine) | Most common FAO disorder (~1 in 15,000); presents at 3–24 months during fasting/illness; hypoketotic hypoglycemia, Reye-like hepatopathy; previously a cause of sudden infant death; ~80% carry common variant c.985A>G (K329E). With NBS and fasting avoidance, prognosis is excellent |
| VLCADD (very long-chain acyl-CoA dehydrogenase deficiency) | VLCAD | ACADVL | Elevated C14:1 | Three phenotypes: severe neonatal (cardiomyopathy, hepatopathy, high mortality), infantile (hypoketotic hypoglycemia), adult (rhabdomyolysis, myopathy) |
| LCHAD (long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency) | LCHAD | HADHA | Elevated C16-OH, C18:1-OH | Hypoketotic hypoglycemia, cardiomyopathy, retinopathy (unique to LCHAD), peripheral neuropathy; maternal HELLP syndrome or AFLP in pregnancies carrying affected fetuses |
| CPT I deficiency | CPT1A | CPT1A | Elevated free carnitine (C0), low C16/C0 ratio | Hypoketotic hypoglycemia, hepatomegaly; primarily hepatic disease; carnitine often paradoxically elevated |
| CPT II deficiency | CPT2 | CPT2 | Elevated C16, C18:1 | Adult myopathic form (most common): exercise/fasting-induced rhabdomyolysis; infantile form: cardiomyopathy, hepatopathy |
General management principles: avoid prolonged fasting (maximum safe fasting time age-dependent: neonates 3–4 hours, infants 6–8 hours, children 10–12 hours); frequent meals; uncooked cornstarch at bedtime for sustained glucose release; low-fat diet (long-chain FAO disorders) with MCT oil supplementation (bypasses long-chain pathway); IV D10 during illness/surgery; carnitine supplementation (for MCADD, controversial for long-chain defects). Emergency protocol for acute decompensation: IV D10 at high rate to suppress lipolysis.
22 Lysosomal Storage Diseases
Lysosomes are membrane-bound organelles containing acid hydrolases that degrade macromolecules. Deficiency of specific lysosomal enzymes leads to substrate accumulation and progressive cellular dysfunction. Most are autosomal recessive (exceptions: Fabry and Hunter — X-linked). Collectively affect ~1 in 5,000–8,000 births.
Mucopolysaccharidoses (MPS)
| Type | Name | Enzyme | Inheritance | Key Features |
|---|---|---|---|---|
| MPS I-H | Hurler | α-L-iduronidase (IDUA) | AR | Most severe MPS I; coarse facies, corneal clouding, dysostosis multiplex, hepatosplenomegaly, cardiac valve disease, intellectual disability. HSCT if <2 years; ERT (laronidase) |
| MPS I-S | Scheie | α-L-iduronidase | AR | Mild form; joint stiffness, corneal clouding, normal intelligence |
| MPS II | Hunter | Iduronate-2-sulfatase (IDS) | X-linked | Similar to Hurler but NO corneal clouding; ivory-colored pebbly skin lesions |
| MPS III | Sanfilippo | 4 subtypes (A-D) | AR | Predominantly neurologic: severe behavioral problems, progressive intellectual disability, mild somatic features |
| MPS IV | Morquio | A: GALNS; B: GLB1 | AR | Severe skeletal dysplasia, short trunk dwarfism, odontoid hypoplasia (cervical instability), corneal clouding, normal intelligence. ERT (elosulfase alfa for MPS IVA) |
| MPS VI | Maroteaux-Lamy | Arylsulfatase B (ARSB) | AR | Severe somatic features (like Hurler), normal intelligence. ERT (galsulfase) |
| MPS VII | Sly | β-glucuronidase (GUSB) | AR | Rare; variable phenotype; hydrops fetalis in severe form. ERT (vestronidase alfa) |
Other Key Lysosomal Storage Diseases
| Disease | Enzyme/Gene | Storage Material | Key Features | Treatment |
|---|---|---|---|---|
| Niemann-Pick A | Acid sphingomyelinase (SMPD1) | Sphingomyelin | Infantile neurovisceral; hepatosplenomegaly, cherry-red spot, progressive neurodegeneration; death by age 3 | Supportive |
| Niemann-Pick B | Acid sphingomyelinase | Sphingomyelin | Visceral (non-neuronopathic); hepatosplenomegaly, interstitial lung disease; survival to adulthood | ERT (olipudase alfa) |
| Niemann-Pick C | NPC1 (95%) or NPC2 | Cholesterol (trafficking defect) | Variable onset; vertical supranuclear gaze palsy (pathognomonic), progressive ataxia, dystonia, cognitive decline, neonatal jaundice/hepatosplenomegaly | Miglustat (substrate reduction) |
| Pompe disease | Acid α-glucosidase (GAA) | Glycogen (lysosomal) | Infantile: severe cardiomyopathy (massive LVH), hypotonia, macroglossia, death by 1 year without ERT. Late-onset: progressive proximal myopathy and respiratory failure | ERT (alglucosidase alfa, avalglucosidase alfa) |
| Krabbe disease | Galactosylceramidase (GALC) | Galactocerebroside | Infantile: irritability, hypertonia, optic atrophy, peripheral neuropathy (elevated CSF protein), "globoid cells" on biopsy; death by age 2 | HSCT (if pre-symptomatic) |
23 Glycogen Storage Diseases
Glycogen storage diseases (GSD) result from defects in glycogen synthesis, degradation, or regulation. Most are autosomal recessive. Classification is by the affected enzyme.
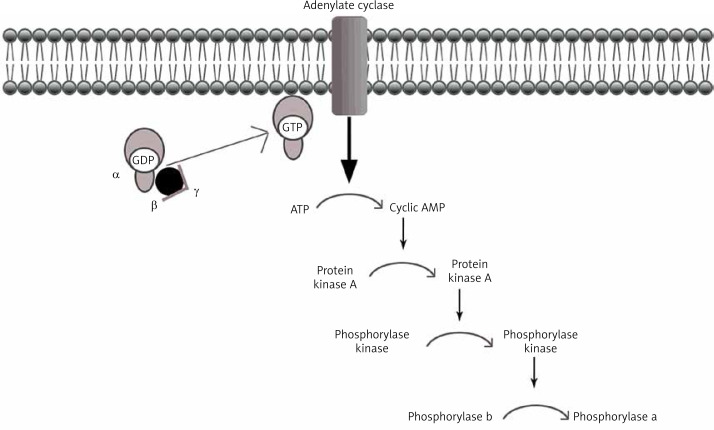
| Type | Name/Enzyme | Gene | Key Features |
|---|---|---|---|
| GSD 0 | Glycogen synthase | GYS2 | Fasting ketotic hypoglycemia (no glycogen to break down); postprandial hyperglycemia and hyperlactatemia |
| GSD Ia | Von Gierke / Glucose-6-phosphatase | G6PC | Most common hepatic GSD; severe fasting hypoglycemia, lactic acidosis, hyperuricemia (gout), hyperlipidemia, hepatomegaly (glycogen and fat), "doll-like" facies, hepatic adenomas (risk of HCC); management: cornstarch, continuous feeds |
| GSD Ib | G6P translocase | SLC37A4 | Same as Ia plus neutropenia and recurrent infections; managed with G-CSF and empagliflozin |
| GSD II | Pompe / Acid maltase (acid α-glucosidase) | GAA | Lysosomal GSD (see section 22); infantile — cardiomyopathy; late-onset — limb-girdle myopathy, respiratory failure. ERT available |
| GSD III | Cori/Forbes / Debranching enzyme | AGL | Hepatomegaly, fasting hypoglycemia and ketosis, elevated CK (myopathy variant IIIa), abnormal glycogen with short outer branches ("limit dextrin"); hepatic symptoms improve with age, muscle disease may progress |
| GSD IV | Andersen / Branching enzyme | GBE1 | Progressive hepatic cirrhosis (amylopectin-like glycogen triggers immune response), failure to thrive; liver transplant |
| GSD V | McArdle / Myophosphorylase | PYGM | Most common muscle GSD; exercise intolerance, myalgias, rhabdomyolysis, "second wind" phenomenon (improved exercise tolerance after rest due to switch to fatty acid oxidation); no rise in lactate on ischemic forearm exercise test |
| GSD VI | Hers / Hepatic phosphorylase | PYGL | Mild hepatomegaly, mild fasting hypoglycemia and ketosis; generally benign course |
| GSD VII | Tarui / Phosphofructokinase (PFK) | PFKM | Similar to McArdle but also compensated hemolysis (PFK in RBCs); exercise intolerance with worsened by glucose/carbohydrate intake ("out of wind" phenomenon) |
| GSD IX | Phosphorylase kinase | PHKA2 (XL), PHKB, PHKG2 | Most common GSD overall; hepatomegaly, mild hypoglycemia, short stature; generally benign, improves with age; X-linked form (IXa) most common |
24 Peroxisomal Disorders & Newborn Screening
Peroxisomal Disorders
Peroxisomes are organelles involved in very long-chain fatty acid (VLCFA) β-oxidation, bile acid synthesis, plasmalogen biosynthesis, and reactive oxygen species metabolism.
Zellweger spectrum disorders (ZSD): peroxisome biogenesis disorders caused by mutations in PEX genes. Three phenotypes of decreasing severity along a spectrum:
- Zellweger syndrome (most severe): neonatal hypotonia, characteristic facies (high forehead, flat supraorbital ridges, wide fontanelles), seizures, hepatomegaly, renal cysts, stippled epiphyses (chondrodysplasia punctata), absent peroxisomes on liver biopsy; death in first year
- Neonatal adrenoleukodystrophy (intermediate): similar but milder features, survival into childhood
- Infantile Refsum disease (mildest): sensorineural hearing loss, retinitis pigmentosa, intellectual disability; survival to adulthood possible
X-linked adrenoleukodystrophy (X-ALD): ABCD1 gene; VLCFA accumulation. Three main phenotypes: childhood cerebral ALD (inflammatory demyelination, rapid neurologic decline, adrenal insufficiency; HSCT can halt disease if done early), adrenomyeloneuropathy (adult males, slowly progressive spasticity and neuropathy), and isolated Addison disease. Diagnosis: elevated plasma VLCFA (C26:0, C26:0/C22:0 ratio). Added to the Recommended Uniform Screening Panel (RUSP) for newborn screening.
Newborn Screening
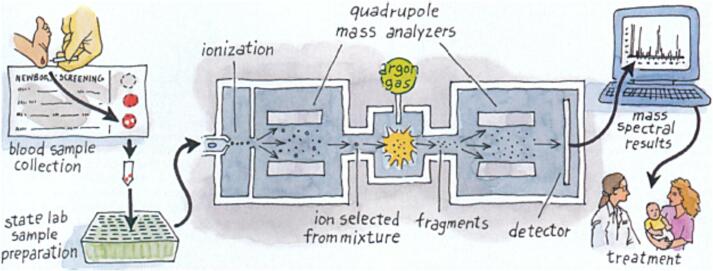
Universal newborn screening (NBS) in the US uses dried blood spots collected at 24–48 hours of life. The Recommended Uniform Screening Panel (RUSP) contains 37 core conditions and 26 secondary conditions (as of 2024). The backbone technology is tandem mass spectrometry (MS/MS), which can simultaneously measure acylcarnitines (for fatty acid oxidation and organic acid disorders) and amino acids from a single dried blood spot.
Amino acid disorders: PKU, MSUD, tyrosinemia type I, homocystinuria, citrullinemia, argininosuccinic aciduria.
Organic acid disorders: propionic acidemia, methylmalonic acidemias, isovaleric acidemia, glutaric acidemia type I, 3-methylcrotonyl-CoA carboxylase deficiency, HMG-CoA lyase deficiency, β-ketothiolase deficiency.
Fatty acid oxidation: MCADD, VLCADD, LCHAD, TFP deficiency, carnitine uptake defect, CPT I and II deficiency.
Hemoglobinopathies: SCD (HbSS, HbSC, HbS/β-thal).
Endocrine: congenital hypothyroidism, congenital adrenal hyperplasia (21-hydroxylase deficiency via 17-OHP).
Other: CF (IRT), galactosemia (GALT enzyme), biotinidase deficiency, severe combined immunodeficiency (SCID, via TREC assay), SMA (SMN1 deletion), Pompe (GAA enzyme), MPS I (IDUA enzyme), X-ALD (C26:0-lysophosphatidylcholine), Krabbe (GALC enzyme, in some states).
Principles of screening: NBS is a screening test, not a diagnostic test — all positive results require confirmatory testing. Sensitivity is prioritized over specificity (false positives are acceptable; false negatives are not). Second screens (repeat at ~2 weeks) performed in some states. Point-of-care screening includes pulse oximetry for critical congenital heart disease and hearing screening (otoacoustic emissions or auditory brainstem response).
25 Hereditary Breast & Ovarian Cancer
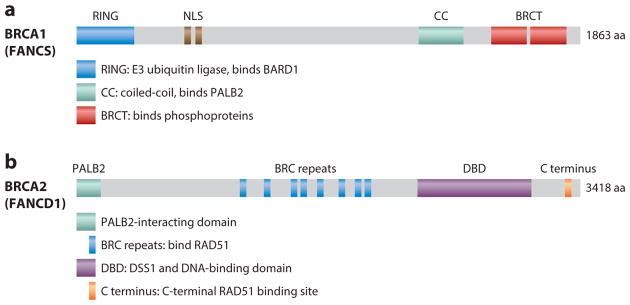
BRCA1 (17q21) and BRCA2 (13q13) encode proteins essential for homologous recombination DNA repair. Autosomal dominant with high penetrance. BRCA1: lifetime breast cancer risk ~55–72%, ovarian cancer ~39–44%. BRCA2: lifetime breast cancer risk ~45–69%, ovarian cancer ~11–17%. BRCA2 also increases risk of male breast cancer, pancreatic cancer, and prostate cancer. Founder mutations are common in Ashkenazi Jewish populations (BRCA1 185delAG, BRCA1 5382insC, BRCA2 6174delT; combined carrier frequency ~1 in 40).
NCCN Management Guidelines (High-Risk Screening & Prevention)
| Intervention | Details |
|---|---|
| Breast cancer screening | Monthly breast self-awareness starting age 18; clinical breast exam every 6–12 months starting age 25; annual breast MRI with contrast age 25–29; annual mammogram + breast MRI age 30–75; individualized >75 years |
| Risk-reducing mastectomy | Option to discuss; reduces breast cancer risk by ~90–95%; consider after age 25 and completion of childbearing |
| Risk-reducing salpingo-oophorectomy (RRSO) | Recommended age 35–40 for BRCA1 (after childbearing), age 40–45 for BRCA2; reduces ovarian cancer risk by ~80%, breast cancer risk by ~50% if done premenopausally |
| Chemoprevention | Tamoxifen or aromatase inhibitors may be considered for breast cancer risk reduction in those who decline surgery |
| Ovarian cancer screening | No effective screening exists; CA-125 and transvaginal ultrasound are not recommended for screening (poor sensitivity/specificity) — this is why RRSO is so strongly recommended |
| BRCA2 prostate cancer | Screening with PSA starting age 40 (males with BRCA2) |
| Pancreatic cancer (BRCA2) | Consider annual MRI/MRCP and endoscopic ultrasound starting age 50 (or 10 years before earliest pancreatic cancer in family) |
Therapeutic implications: BRCA-mutated cancers are sensitive to platinum chemotherapy and PARP inhibitors (olaparib, rucaparib, niraparib, talazoparib) via synthetic lethality (HR-deficient cells cannot repair PARP-inhibitor-induced DNA damage).
Other high-risk genes for breast cancer: PALB2 (partner and localizer of BRCA2; lifetime breast cancer risk ~33–58%), TP53 (Li-Fraumeni), PTEN (Cowden syndrome), CDH1 (hereditary diffuse gastric cancer + lobular breast cancer), STK11 (Peutz-Jeghers), ATM and CHEK2 (moderate-risk genes, ~2× relative risk).
NCCN Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic26 Lynch Syndrome & GI Cancer Genetics
Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer — HNPCC)
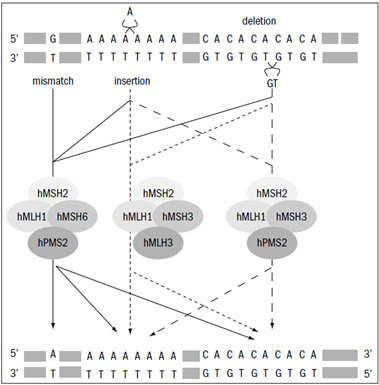
Autosomal dominant; caused by germline mutations in DNA mismatch repair (MMR) genes: MLH1 (most common), MSH2, MSH6, PMS2, or EPCAM (deletions silencing MSH2). Most common hereditary colorectal cancer syndrome (~3–5% of all CRC). Lifetime CRC risk: ~40–80% (MLH1/MSH2), ~10–22% (MSH6), ~15–20% (PMS2). Also increases risk of endometrial cancer (~25–60%), ovarian (~4–24%), gastric, urinary tract, small bowel, hepatobiliary, and sebaceous neoplasms.
Diagnostic Criteria
≥3 relatives with Lynch-associated cancer AND ≥2 successive generations AND ≥1 diagnosed before age 50 AND 1 is first-degree relative of the other two AND FAP excluded. Highly specific but low sensitivity (~40%).
Revised Bethesda Guidelines (for selecting tumors for MSI/IHC testing): CRC diagnosed <50; synchronous/metachronous Lynch-associated tumors; CRC with MSI-H histology <60; CRC with ≥1 first-degree relative with Lynch tumor <50; CRC with ≥2 first/second-degree relatives with Lynch tumor at any age.
Universal tumor screening (now recommended): all CRCs tested by immunohistochemistry (IHC) for MLH1, MSH2, MSH6, PMS2 protein expression AND/OR microsatellite instability (MSI) testing. Loss of MLH1/PMS2 → test for MLH1 promoter methylation and BRAF V600E (if both positive, likely sporadic; if negative, germline MLH1 testing). Loss of MSH2/MSH6 or MSH6 alone or PMS2 alone → proceed directly to germline testing.
Lynch Syndrome Surveillance
- Colonoscopy: every 1–2 years starting age 20–25 (or 2–5 years before youngest CRC in family)
- Endometrial cancer: education about symptoms; consider endometrial sampling annually starting age 30–35; discuss risk-reducing hysterectomy + BSO after childbearing (especially MLH1/MSH2)
- Aspirin: 600 mg daily shown to reduce CRC incidence (CAPP2 trial); optimal dose under study (ongoing CAPP3 trial at 100 mg, 300 mg, 600 mg)
- Immunotherapy: MSI-H/dMMR tumors respond dramatically to immune checkpoint inhibitors (pembrolizumab approved first-line for MSI-H CRC; dostarlimab for dMMR rectal cancer — 100% complete clinical response in initial trial)
Familial Adenomatous Polyposis (FAP)
Autosomal dominant; APC gene (5q21). Classic FAP: >100 colorectal adenomatous polyps by teens–20s; ~100% CRC risk by age 40 without colectomy. Attenuated FAP: 10–100 polyps, later onset, lower (but still elevated) CRC risk. Gardner syndrome: FAP + desmoid tumors, osteomas (mandible, skull), epidermoid cysts, dental anomalies. Turcot syndrome: FAP + CNS tumors (medulloblastoma in APC-related Turcot; glioblastoma in Lynch-related Turcot). Management: prophylactic total colectomy (proctocolectomy with IPAA) typically in late teens to 20s; upper GI surveillance (periampullary adenomas → duodenal cancer risk).
Hereditary Diffuse Gastric Cancer
Autosomal dominant; CDH1 gene (E-cadherin). Lifetime gastric cancer risk ~70% (diffuse/signet ring type); lobular breast cancer risk in women ~40–50%. Screening endoscopy has poor sensitivity for diffuse gastric cancer (tumor grows beneath mucosa). IGCLC guidelines recommend prophylactic total gastrectomy at age 20–30 for confirmed pathogenic variant carriers.
27 MEN Syndromes, VHL & Other Cancer Predisposition
Multiple Endocrine Neoplasia (MEN) Syndromes
| Syndrome | Gene | Inheritance | Components |
|---|---|---|---|
| MEN1 (Wermer syndrome) | MEN1 (menin, tumor suppressor) | AD | 3 P's: (1) Parathyroid adenomas (~90%, primary hyperparathyroidism — usually first manifestation); (2) Pancreatic/duodenal neuroendocrine tumors (gastrinoma — Zollinger-Ellison, insulinoma); (3) Pituitary adenomas (~40%, prolactinoma most common). Also: adrenal cortical adenomas, carcinoid tumors (thymic, bronchial), facial angiofibromas, collagenomas |
| MEN2A (Sipple syndrome) | RET proto-oncogene (gain of function) | AD | (1) Medullary thyroid carcinoma (MTC, ~95% — virtually 100% penetrance); (2) Pheochromocytoma (~50%, often bilateral); (3) Primary hyperparathyroidism (~20–30%). Specific RET codon mutations correlate with risk level (ATA risk categories: highest, high, moderate) |
| MEN2B | RET (M918T in ~95%) | AD | (1) MTC (earliest onset, most aggressive); (2) Pheochromocytoma (~50%); (3) Mucosal neuromas (lips, tongue, eyelids — characteristic "bumpy lips"); (4) Intestinal ganglioneuromatosis; (5) Marfanoid habitus. No hyperparathyroidism. ~75% de novo |
RET testing and prophylactic thyroidectomy: MEN2B (highest risk) — thyroidectomy within first 6 months of life; MEN2A high risk (C634R) — by age 5; MEN2A moderate risk — may delay if calcitonin normal, but usually by teens. Annual screening for pheochromocytoma (plasma/urine metanephrines) and hyperparathyroidism (calcium, PTH) starting in childhood.
Von Hippel-Lindau Disease (VHL)
Autosomal dominant; VHL gene (3p25.3), a tumor suppressor involved in HIF (hypoxia-inducible factor) degradation. Loss of VHL → constitutive HIF activation → VEGF overproduction → highly vascular tumors. Components: CNS hemangioblastomas (cerebellum > spinal cord > brainstem, ~60–80%), retinal hemangioblastomas (~60%), clear cell renal cell carcinoma (~40%, bilateral/multifocal, leading cause of death), pheochromocytoma (~10–20%), pancreatic neuroendocrine tumors (~15%), endolymphatic sac tumors (hearing loss), pancreatic cysts, epididymal cystadenomas. Surveillance: annual ophthalmologic exam, annual plasma metanephrines, abdominal MRI/ultrasound, brain/spine MRI every 2 years — all starting in childhood/adolescence. Nephron-sparing surgery for renal tumors (to preserve renal function given multifocality). Belzutifan (HIF-2α inhibitor) approved for VHL-associated RCC, hemangioblastomas, and pNET.
Li-Fraumeni Syndrome
Autosomal dominant; TP53 germline mutation. p53 is the "guardian of the genome" — master regulator of DNA repair, cell cycle arrest, and apoptosis. Extremely high cancer risk: ~50% by age 30, >90% lifetime. Core tumors (SBLA): Sarcomas, Breast cancer (premenopausal), Leukemia/lymphoma, Adrenal cortical carcinoma (childhood), brain tumors (especially choroid plexus carcinoma). Also: lung, GI, thyroid, gonadal, many others. Surveillance: annual whole-body MRI, brain MRI, breast MRI, colonoscopy, dermatologic exam, and others (Toronto protocol). Avoid radiation exposure when possible (increased susceptibility to radiation-induced malignancy).
Retinoblastoma
Caused by loss of function of RB1 (13q14), the prototypical tumor suppressor gene. Knudson two-hit hypothesis: bilateral/hereditary retinoblastoma requires one germline mutation ("first hit," present in all cells) + one somatic mutation ("second hit," loss of remaining allele in retinal cell); sporadic retinoblastoma requires two somatic mutations in the same cell (hence unilateral and later onset). Hereditary form (~40%): bilateral, multifocal, mean age 12 months; also predisposes to osteosarcoma, soft tissue sarcomas, melanoma, and other cancers in adulthood. Sporadic form (~60%): unilateral, unifocal, mean age 24 months. Management: enucleation, chemotherapy, focal therapies (laser, cryotherapy); genetic counseling for all families.
28 Tumor Suppressor Genes vs Oncogenes
Understanding the two major classes of cancer genes is fundamental to cancer genetics.
| Feature | Tumor Suppressor Genes | Oncogenes |
|---|---|---|
| Normal function | Inhibit cell growth, promote apoptosis, repair DNA ("brakes") | Promote cell growth, proliferation, survival ("accelerator") |
| Mutation effect | Loss of function | Gain of function |
| Alleles needed | Both alleles must be inactivated (two-hit model) | Only one allele needs to be mutated (dominant) |
| Inheritance in cancer syndromes | One hit germline, second hit somatic | Germline gain-of-function mutations (rare) |
| Examples | RB1, TP53, APC, BRCA1, BRCA2, VHL, MLH1, MSH2, PTEN, WT1, NF1, NF2, SMAD4 | RET, MET, HER2, RAS (KRAS, NRAS, HRAS), MYC, BCR-ABL, BRAF, ALK, EGFR, KIT, JAK2 |
| Mechanisms of inactivation/activation | Deletion, point mutation, promoter methylation, LOH | Point mutation, amplification, translocation creating fusion gene |
BCR-ABL: t(9;22) Philadelphia chromosome in CML → constitutively active tyrosine kinase → treated with imatinib.
RAS mutations: KRAS (codon 12, 13, 61) in ~25% of all human cancers (pancreatic, CRC, NSCLC).
BRAF V600E: melanoma (~50%), thyroid cancer, hairy cell leukemia → vemurafenib/dabrafenib.
HER2 amplification: breast cancer (~20%) → trastuzumab.
MYC amplification: neuroblastoma (MYCN), Burkitt lymphoma (MYC translocation t(8;14)).
29 Carrier Screening & Prenatal Screening
Carrier Screening
ACOG recommends offering carrier screening to all pregnant individuals (or those planning pregnancy). Two approaches:
- Ethnicity-based screening (traditional): targeted to high-prevalence conditions in specific populations — CF (all), SCD/hemoglobinopathies (African, Mediterranean, SE Asian), Tay-Sachs (Ashkenazi Jewish, French-Canadian, Cajun), β-thalassemia (Mediterranean, SE Asian, African), Canavan disease and familial dysautonomia (Ashkenazi Jewish)
- Expanded carrier screening (ECS) (increasingly standard): pan-ethnic panels testing 100–400+ conditions simultaneously using NGS; identifies carriers of conditions that ethnicity-based screening would miss; ACOG supports offering ECS to all patients regardless of ethnicity
Prenatal Screening for Aneuploidy
| Test | Timing | Components | Detection Rate for Trisomy 21 |
|---|---|---|---|
| First trimester combined screen | 11–13+6 weeks | Nuchal translucency (NT) ultrasound + serum PAPP-A (low) + free β-hCG (high) | ~82–87% (5% FPR) |
| Quad screen | 15–22 weeks | AFP (low), hCG (high), uE3 (low), inhibin A (high) | ~81% (5% FPR) |
| Integrated screen | First + second trimester combined | First trimester (NT + PAPP-A) + second trimester (quad screen) — single result | ~95% (1% FPR) |
| Cell-free DNA (cfDNA/NIPT) | ≥10 weeks | Analysis of fetal DNA fragments in maternal blood; evaluates trisomies 21, 18, 13, sex chromosome aneuploidies, and some microdeletions | ~99% for T21 (0.1% FPR); ~97% for T18; ~91% for T13 |
cfDNA is a screening test, not a diagnostic test. Despite high sensitivity and specificity, the positive predictive value (PPV) depends on the prevalence of the condition in the screened population. PPV for T21 is ~90% in high-risk populations but may be ~50% in low-risk populations. All positive cfDNA results must be confirmed by diagnostic testing (CVS or amniocentesis). cfDNA performance is reduced in: obesity (lower fetal fraction), early GA (<10 weeks), mosaicism, vanishing twin, and maternal copy number variants.
Serum marker patterns (useful for boards):
| Condition | AFP | hCG | uE3 | Inhibin A |
|---|---|---|---|---|
| Trisomy 21 | ↓ | ↑ | ↓ | ↑ |
| Trisomy 18 | ↓ | ↓ | ↓ | ↓ |
| Open NTD | ↑↑ | Normal | Normal | Normal |
| Abdominal wall defect | ↑↑ | Normal | Normal | Normal |
30 Prenatal Diagnostic Testing & PGT
Invasive Prenatal Diagnostic Tests
| Feature | Chorionic Villus Sampling (CVS) | Amniocentesis |
|---|---|---|
| Timing | 10–13 weeks | 15–20 weeks (typically 16–18) |
| Specimen | Placental trophoblast (chorionic villi) | Amniotic fluid (fetal cells — desquamated skin, urinary tract, respiratory epithelium) |
| Technique | Transcervical or transabdominal needle/catheter | Transabdominal needle aspiration under ultrasound guidance |
| Procedure-related pregnancy loss | ~0.1–0.2% (similar to amniocentesis) | ~0.1–0.3% |
| Karyotype results | Preliminary (direct prep): 2–3 days; final (culture): 7–14 days | 10–14 days (culture) |
| Limitations | Confined placental mosaicism (CPM) in ~1–2% — may not reflect true fetal genotype; cannot test for NTDs (no AFP available) | Rare risk of amniotic fluid leak, chorioamnionitis |
| Additional tests available | Karyotype, FISH, CMA, single-gene testing, biochemical testing | Same + amniotic fluid AFP and acetylcholinesterase for NTDs |
Preimplantation Genetic Testing (PGT)
Performed during in vitro fertilization (IVF) by biopsy of trophectoderm cells from day 5–7 blastocysts. Three types:
- PGT-M (monogenic/single-gene disorders): tests for a specific known familial mutation (e.g., CF, SCD, Huntington disease). Requires prior probe development from family samples
- PGT-A (aneuploidy): screens for whole-chromosome gains/losses. Used to improve IVF success rates by selecting euploid embryos, particularly in advanced maternal age, recurrent miscarriage, or recurrent implantation failure. Controversial — not universally recommended
- PGT-SR (structural rearrangements): for carriers of balanced translocations or inversions who are at risk of producing unbalanced embryos
31 Teratology & Genetic Counseling
Teratology
A teratogen is any agent that causes structural or functional abnormalities in the developing embryo/fetus. The effect depends on: timing of exposure (critical period), dose, genetic susceptibility, and the specific agent.
Critical periods: weeks 3–8 of gestation (embryonic period) is the time of greatest susceptibility to major structural malformations (organogenesis). Before week 3 — "all-or-none" effect (either embryo dies or survives without defect). After week 8 (fetal period) — exposure mainly causes functional defects and growth restriction rather than major structural anomalies, though some organs (e.g., brain, genitalia) continue to be vulnerable.
Key Teratogens
| Teratogen | Effects | Category/Notes |
|---|---|---|
| Isotretinoin (Accutane) | Craniofacial (microtia, cleft palate), cardiac (conotruncal), CNS (hydrocephalus, microcephaly), thymic hypoplasia | Most teratogenic drug; contraception required 1 month before, during, and 1 month after use (iPLEDGE program) |
| Valproic acid | Neural tube defects (1–2% risk, especially spina bifida), craniofacial, cognitive impairment, cardiac defects, hypospadias | Avoid in pregnancy if possible; folic acid 4–5 mg/day may reduce NTD risk |
| Warfarin | First trimester: nasal hypoplasia, stippled epiphyses (warfarin embryopathy); second/third trimester: CNS abnormalities, hemorrhage | Switch to LMWH or unfractionated heparin for first trimester; warfarin safest in 2nd trimester for high-risk mechanical valves |
| ACE inhibitors/ARBs | Second/third trimester: renal agenesis/dysgenesis, oligohydramnios (Potter sequence), skull ossification defects, IUGR | Contraindicated in pregnancy; switch to labetalol, nifedipine, or methyldopa |
| Thalidomide | Phocomelia (limb reduction), cardiac, GI, renal anomalies | Exposure during days 20–36 post-conception; strict pregnancy prevention (REMS program) |
| Alcohol | Fetal alcohol spectrum disorders (FASD): smooth philtrum, thin upper lip, short palpebral fissures, microcephaly, intellectual disability, cardiac defects (VSD, ASD), growth restriction | No safe level of alcohol in pregnancy; most common preventable cause of intellectual disability |
| Lithium | Ebstein anomaly (apical displacement of tricuspid valve); risk lower than previously estimated (~0.1–0.2%) | Fetal echocardiography recommended; benefits may outweigh risks in severe bipolar disorder |
| Methotrexate | Aminopterin syndrome: cranial dysostosis, cleft palate, limb anomalies, growth restriction | Absolutely contraindicated; wait ≥3 months after discontinuation before conception |
TORCH Infections
| Infection | Fetal/Neonatal Effects | Distinguishing Features |
|---|---|---|
| Toxoplasmosis | Intracranial calcifications (diffuse/scattered), hydrocephalus, chorioretinitis | Calcifications are diffuse (vs periventricular in CMV) |
| Rubella | Sensorineural hearing loss (most common), cataracts, cardiac defects (PDA, pulmonary stenosis), "blueberry muffin" rash (extramedullary hematopoiesis) | Highest risk if infection in first 12 weeks |
| CMV | Most common congenital infection (~0.5–1%); periventricular calcifications, microcephaly, sensorineural hearing loss, hepatosplenomegaly, petechiae, chorioretinitis | Periventricular calcifications (vs diffuse in toxo) |
| HSV | Neonatal herpes: skin vesicles, encephalitis, disseminated infection; peripartum transmission during vaginal delivery (primary > recurrent) | C-section if active genital lesions at delivery |
| Syphilis | Hydrops, hepatosplenomegaly, rhinitis ("snuffles"), osteochondritis, rash; late: Hutchinson teeth, interstitial keratitis, saber shins | Treatable with penicillin; screen all pregnancies |
| Varicella (VZV) | Congenital varicella syndrome (if <20 weeks): limb hypoplasia, cicatricial skin lesions, eye anomalies, CNS defects | Risk ~0.4–2% if maternal infection in first 20 weeks |
| Zika virus | Microcephaly (severe), brain disruption, arthrogryposis, eye anomalies, sensorineural hearing loss | Congenital Zika syndrome; mosquito-borne or sexual transmission |
Genetic Counseling Principles
Genetic counseling is the process of helping individuals and families understand and adapt to the medical, psychological, and familial implications of genetic contributions to disease (NSGC definition). Core principles include:
- Non-directive approach: providing information and support without making recommendations for reproductive decisions; respecting patient autonomy
- Risk assessment: calculation of recurrence risks based on inheritance pattern, family history, Bayesian analysis, and genetic test results
- Informed consent: thorough discussion of benefits, risks, limitations, and possible outcomes of genetic testing before testing is performed
- Psychosocial support: addressing grief, guilt, anxiety, and family dynamics related to genetic diagnoses
- Privacy and confidentiality: GINA (Genetic Information Nondiscrimination Act) protects against discrimination by health insurers and employers based on genetic information; does NOT cover life, disability, or long-term care insurance
32 Cytogenetic & Molecular Cytogenetic Testing
Conventional Karyotype (G-banding)
Gold standard cytogenetic test. Cells are cultured (from blood, amniotic fluid, or tissue), arrested in metaphase with colchicine, stained with Giemsa to produce characteristic banding patterns. Resolution: ~5–10 Mb (can detect deletions, duplications, and rearrangements only if they are large enough to alter the banding pattern). Detects: numerical abnormalities (aneuploidies, polyploidy), large structural rearrangements (translocations, inversions, large deletions/duplications, ring chromosomes, isochromosomes). Turnaround time: 7–14 days (requires cell culture). Limitations: cannot detect submicroscopic CNVs (<5 Mb), balanced rearrangements may appear normal, point mutations are invisible.
Fluorescence in situ Hybridization (FISH)
Uses fluorescently labeled DNA probes that hybridize to specific chromosome regions. Can be performed on metaphase or interphase cells (no culture needed for interphase FISH — results in 24–48 hours). Resolution: ~100 kb–1 Mb depending on probe size.
| Probe Type | Application | Example |
|---|---|---|
| Centromeric (alpha-satellite) | Enumerate chromosomes (detect aneuploidy) | Rapid interphase FISH for trisomies 13, 18, 21, X, Y on uncultured amniocytes |
| Locus-specific | Detect microdeletions/microduplications at specific loci | 22q11.2 deletion (DiGeorge), 7q11.23 deletion (Williams), 15q11-13 (PWS/Angelman), BCR-ABL fusion |
| Subtelomeric | Screen for subtelomeric rearrangements (cause ~5% of unexplained ID) | Subtelomeric FISH panel (now largely replaced by CMA) |
| Whole chromosome paint | Identify material of unknown chromosomal origin (marker chromosomes, complex rearrangements) | Painting entire chromosome 22 to characterize a derivative chromosome |
Chromosomal Microarray (CMA)
Now recommended as a first-tier test (over karyotype) for patients with intellectual disability, developmental delay, autism spectrum disorder, and multiple congenital anomalies (ACMG 2010 guideline). Resolution: ~50–100 kb (50–100× higher than karyotype). Two main platforms:
| Feature | Array CGH (aCGH) | SNP Array |
|---|---|---|
| Technology | Compares patient DNA to reference DNA by competitive hybridization to genomic probes | Hybridizes patient DNA to probes containing known SNPs |
| Detects CNVs | Yes (deletions and duplications) | Yes |
| Detects LOH/UPD | No | Yes (long contiguous stretches of homozygosity — absence of heterozygosity) |
| Detects triploidy | No (ratios cancel out) | Yes |
| Detects consanguinity | No | Yes (multiple regions of homozygosity across genome) |
Diagnostic yield of CMA: ~15–20% in developmental delay/ID (compared with ~3% for karyotype). However, CMA cannot detect balanced rearrangements (reciprocal translocations, inversions) or low-level mosaicism (<20%). Variants of uncertain significance (VOUS/VUS) are found in ~5–10% of cases — parental testing and database searches help reclassify over time.
33 Molecular Genetic Testing (Sequencing)
Sanger Sequencing
The original DNA sequencing method (chain termination). Reads one gene (or one exon) at a time. Resolution: single nucleotide. Used for: confirmatory testing, targeted testing of known familial variants, sequencing of small genes, and validation of NGS findings. Limitations: labor-intensive and expensive for multi-gene analysis; cannot detect large deletions/duplications (use MLPA for those).
Next-Generation Sequencing (NGS) Panels
Massively parallel sequencing of many genes simultaneously. Gene panels are curated lists of genes associated with a specific phenotype (e.g., cardiomyopathy panel: 50–100+ genes; hereditary cancer panel: 50–80+ genes; epilepsy panel: 200+ genes; RASopathy panel). Advantages: faster and cheaper per gene than Sanger; high depth of coverage (>100×). Limitations: limited to selected genes; may miss genes not on the panel; VUS are common.
Whole Exome Sequencing (WES)
Sequences all ~22,000 protein-coding genes (the exome — ~1.5% of the genome, but contains ~85% of known disease-causing mutations). Diagnostic yield: ~25–40% for undiagnosed genetic conditions (higher in consanguineous families, ~50–60%). Trio analysis (proband + both parents) greatly improves interpretation by identifying de novo mutations and defining phase (cis/trans). Increasingly used as a first-line test for undiagnosed suspected genetic conditions after negative CMA and targeted testing.
Whole Genome Sequencing (WGS)
Sequences the entire genome, including non-coding regions. Advantages over WES: detects structural variants, intronic variants affecting splicing, mitochondrial DNA mutations, and provides uniform coverage. Diagnostic yield: ~5–10% higher than WES in some studies. Limitations: enormous data volume, many VUS, higher cost, longer analysis time, incidental findings in non-coding regions.
RNA Sequencing (RNA-seq)
Complements DNA sequencing by analyzing gene expression, alternative splicing, and fusion transcripts. Particularly useful for detecting splicing abnormalities caused by intronic or synonymous variants that are difficult to interpret from DNA sequence alone. Emerging as a tool for resolving VUS and increasing diagnostic yield in patients negative by WES.
Other Specialized Tests
- Methylation analysis: detects epigenetic changes; essential for diagnosing Prader-Willi/Angelman syndromes, Beckwith-Wiedemann, and imprinting disorders; Southern blot or methylation-specific PCR/MLPA
- Trinucleotide repeat testing: PCR-based or Southern blot for specific repeat expansions (Fragile X CGG, Huntington CAG, myotonic dystrophy CTG, Friedreich ataxia GAA, SCA CAG); standard sequencing cannot reliably size large expansions
- MLPA (multiplex ligation-dependent probe amplification): detects deletions/duplications of individual exons; essential for DMD/BMD, SMA (SMN1 copy number), and other conditions where intragenic deletions/duplications are common
34 Pharmacogenomics
Pharmacogenomics studies how genetic variation affects drug response, including efficacy and toxicity. Implementation of pharmacogenomic testing can optimize drug selection and dosing, reduce adverse drug reactions, and improve clinical outcomes. The Clinical Pharmacogenetics Implementation Consortium (CPIC) provides evidence-based guidelines.
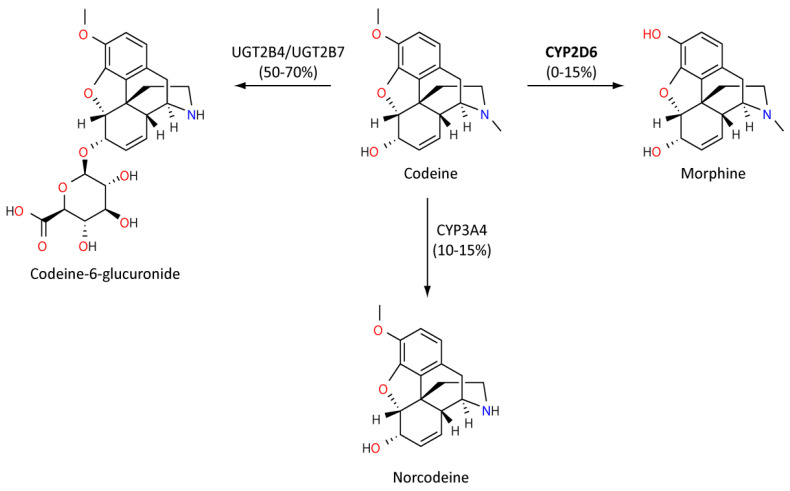
| Gene | Drug(s) | Clinical Application |
|---|---|---|
| CYP2D6 | Codeine, tramadol, tamoxifen, SSRIs, TCAs, ondansetron, many others | Ultra-rapid metabolizers: risk of toxicity from codeine (rapid conversion to morphine → respiratory depression, especially in children); poor metabolizers: codeine ineffective, tamoxifen may be less effective (cannot convert to active metabolite endoxifen). ~6–10% of Caucasians are poor metabolizers; ~1–2% ultra-rapid |
| CYP2C19 | Clopidogrel, PPIs, voriconazole, SSRIs | Poor metabolizers (~2–15%, higher in East Asian): cannot activate clopidogrel (prodrug) → increased risk of stent thrombosis; prasugrel or ticagrelor preferred. Ultra-rapid metabolizers: may need higher PPI doses; lower voriconazole levels |
| HLA-B*5701 | Abacavir (HIV NRTI) | Mandatory testing before abacavir use. HLA-B*5701 positive (~5–8% of Caucasians): risk of severe hypersensitivity reaction (fever, rash, GI symptoms, potentially fatal rechallenge); absolute contraindication to abacavir. One of the most successful pharmacogenomic implementations |
| HLA-B*1502 | Carbamazepine, oxcarbazepine | High prevalence in SE Asian populations (~8%). Positive: risk of Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN). FDA recommends testing before use in at-risk populations |
| HLA-B*5801 | Allopurinol | Risk of severe SJS/TEN and DRESS. Higher prevalence in Korean, Southeast Asian, and African American populations. Consider testing before initiating allopurinol, especially in at-risk populations |
| TPMT / NUDT15 | Azathioprine, 6-mercaptopurine, thioguanine | Poor metabolizers: risk of severe myelosuppression (life-threatening pancytopenia) due to accumulation of active thioguanine nucleotides. ~10% are intermediate metabolizers (dose reduce), ~0.3% poor metabolizers (avoid thiopurines or use 10% of standard dose). NUDT15 especially relevant in East Asian populations |
| DPYD | 5-fluorouracil, capecitabine | DPD deficiency (partial in ~3–8%, complete in ~0.1–0.5%): risk of severe toxicity (neutropenia, mucositis, diarrhea, neurotoxicity); DPYD*2A most common variant; CPIC recommends reduced dose for intermediate metabolizers, avoidance for poor metabolizers |
| VKORC1 + CYP2C9 | Warfarin | VKORC1 variants affect vitamin K epoxide reductase sensitivity; CYP2C9 variants reduce warfarin metabolism. FDA-approved pharmacogenomic dosing algorithms incorporate these variants along with clinical factors |
| CYP3A5 | Tacrolimus | Expressors (CYP3A5*1 carriers): require higher tacrolimus doses to achieve target levels; non-expressors: standard dosing |
| SLCO1B1 | Simvastatin | SLCO1B1*5 (rs4149056 C allele): reduced hepatic uptake → increased plasma statin levels → higher risk of myopathy/rhabdomyolysis; avoid simvastatin 80 mg; consider alternative statins or lower doses |
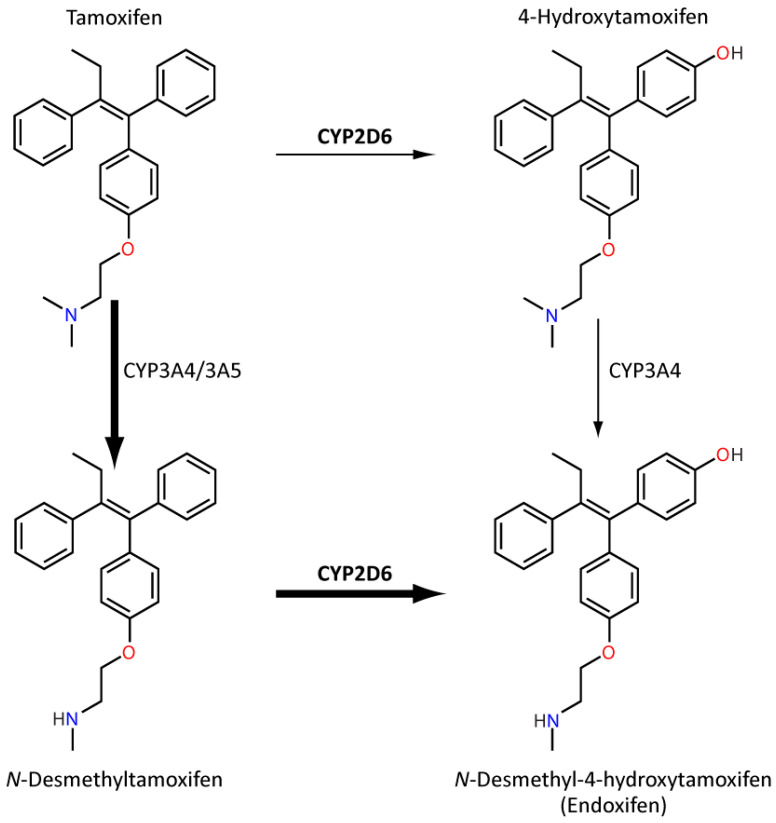
35 ACMG Variant Classification & Pedigree Symbols
ACMG/AMP Variant Classification (2015 Standards and Guidelines)
The American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) established a five-tier classification system for sequence variants, using defined criteria with weighted evidence categories.
| Classification | Definition | Clinical Action |
|---|---|---|
| Pathogenic (P) | Strong evidence that the variant causes disease | Report and use for clinical decision-making, predictive testing in family members, and prenatal diagnosis |
| Likely Pathogenic (LP) | >90% probability of being pathogenic | May be used for clinical decision-making (same as pathogenic in most guidelines); confirm with additional evidence if possible |
| Variant of Uncertain Significance (VUS) | Insufficient evidence to classify as pathogenic or benign | Do NOT use for clinical decision-making; may be reclassified over time with additional data; recommend periodic reanalysis; parental/family testing may help clarify |
| Likely Benign (LB) | >90% probability of being benign | Generally not reported or reported without clinical concern |
| Benign (B) | Strong evidence that the variant does not cause disease | Generally not reported |
Pathogenic evidence:
PVS1 (very strong): null variant in a gene where loss of function is a known mechanism (nonsense, frameshift, canonical splice site, exon deletion).
PS1 (strong): same amino acid change as a previously established pathogenic variant.
PS2 (strong): de novo (confirmed) in a patient with disease and no family history.
PS3 (strong): well-established functional studies showing damaging effect.
PM1 (moderate): located in a critical functional domain (hot spot).
PM2 (moderate): absent from population databases (gnomAD).
PP1 (supporting): cosegregation with disease in family.
PP3 (supporting): multiple computational algorithms predict deleterious effect.
Benign evidence:
BA1 (stand-alone): allele frequency >5% in any population database.
BS1 (strong): allele frequency greater than expected for disorder.
BS3 (strong): well-established functional studies showing no damaging effect.
BP1 (supporting): missense variant in a gene where truncating variants cause disease.
BP4 (supporting): computational evidence suggests no impact.
Combining criteria: Pathogenic requires ≥1 very strong + ≥1 strong (or other combinations); likely pathogenic requires less evidence; likely benign requires ≥1 strong + ≥1 supporting benign; benign requires ≥1 stand-alone OR ≥2 strong.
Standard Pedigree Symbols
Pedigree construction follows standardized nomenclature recommended by the National Society of Genetic Counselors (NSGC) and the Pedigree Standardization Work Group.
| Symbol | Meaning |
|---|---|
| □ (Square) | Male |
| ○ (Circle) | Female |
| ◇ (Diamond) | Sex unspecified or unknown |
| ■ / ● (Filled) | Affected individual |
| Half-filled (upper/lower/left/right) | Carrier or partial expression (specify in legend) |
| Dot inside symbol | Obligate carrier (deduced from pedigree analysis) |
| Diagonal line through symbol | Deceased |
| Arrow with "P" | Proband (index case) |
| Horizontal line between symbols | Mating/relationship |
| Double horizontal line | Consanguineous mating |
| Vertical line down from mating line | Offspring |
| Roman numerals (I, II, III) | Generations |
| Arabic numerals (1, 2, 3) | Individuals within a generation (left to right) |
| Triangle | Pregnancy or miscarriage/spontaneous abortion |
| "SB" inside symbol | Stillbirth |
| "P" inside diamond/triangle | Current pregnancy |
| Adoption: brackets around symbol | Adopted into family (brackets in) or out of family (brackets out) |
36 Genetic Testing Algorithm by Clinical Scenario
| Clinical Scenario | First-Line Test | Second-Line / Follow-Up |
|---|---|---|
| Suspected aneuploidy (e.g., Down syndrome features) | Karyotype (G-banding) | FISH for rapid confirmation; CMA if mosaic suspected |
| Unexplained intellectual disability / developmental delay / ASD | Chromosomal microarray (CMA) + Fragile X testing | Gene panels or WES if CMA/FraX negative; consider metabolic workup |
| Multiple congenital anomalies / dysmorphic features | CMA | Targeted gene testing based on pattern; WES if nondiagnostic |
| Suspected single-gene disorder (e.g., CF, SCD) | Targeted gene testing (Sanger or NGS) | MLPA for deletions/duplications if sequencing negative |
| Suspected microdeletion syndrome | CMA or targeted FISH | CMA preferred (detects both known and novel CNVs) |
| Recurrent pregnancy loss | Parental karyotypes (balanced translocation?) | CMA on products of conception |
| Hereditary cancer predisposition | Multi-gene NGS panel | Consider tumor testing (somatic) if germline negative but clinical suspicion high |
| Suspected metabolic disorder | Targeted metabolite analysis (plasma amino acids, urine organic acids, acylcarnitine profile) | Enzyme assay in fibroblasts/leukocytes; molecular confirmation |
| Prenatal: positive screening for aneuploidy | CVS (10–13 wk) or amniocentesis (15–20 wk) for karyotype ± CMA | FISH for rapid result while awaiting culture |
| Undiagnosed after initial workup | Whole exome sequencing (trio preferred) | Whole genome sequencing; RNA-seq; reanalysis of existing data |
| Imprinting disorder suspected (PWS, AS, BWS) | Methylation analysis | FISH or CMA for deletion; UPD studies; gene sequencing (UBE3A for AS) |
| Trinucleotide repeat disorder suspected | Repeat-primed PCR or Southern blot for specific expansion | Standard sequencing cannot reliably size large expansions |
37 Medications in Genetics
Enzyme Replacement Therapy (ERT)
| Disease | Drug | Target/Enzyme Replaced | Route / Key Notes |
|---|---|---|---|
| Gaucher type I | Imiglucerase (Cerezyme), velaglucerase alfa (VPRIV), taliglucerase alfa (Elelyso) | Glucocerebrosidase | IV every 2 weeks; dramatic improvement in visceral and hematologic disease; does not cross BBB (no CNS benefit) |
| Fabry disease | Agalsidase alfa (Replagal), agalsidase beta (Fabrazyme) | α-Galactosidase A | IV every 2 weeks; slows renal and cardiac progression |
| Pompe disease | Alglucosidase alfa (Lumizyme), avalglucosidase alfa (Nexviazyme) | Acid α-glucosidase (GAA) | IV every 2 weeks; life-saving in infantile form; Nexviazyme has enhanced uptake |
| MPS I (Hurler/Scheie) | Laronidase (Aldurazyme) | α-L-iduronidase | IV weekly; does not cross BBB; HSCT preferred for severe Hurler if <2 years |
| MPS II (Hunter) | Idursulfase (Elaprase) | Iduronate-2-sulfatase | IV weekly |
| MPS IVA (Morquio A) | Elosulfase alfa (Vimizim) | N-acetylgalactosamine-6-sulfatase | IV weekly |
| MPS VI (Maroteaux-Lamy) | Galsulfase (Naglazyme) | Arylsulfatase B | IV weekly |
| MPS VII (Sly) | Vestronidase alfa (Mepsevii) | β-Glucuronidase | IV every 2 weeks |
| Niemann-Pick B | Olipudase alfa (Xenpozyme) | Acid sphingomyelinase | IV every 2 weeks; dose escalation required |
| CLN2 (neuronal ceroid lipofuscinosis type 2) | Cerliponase alfa (Brineura) | TPP1 | Intracerebroventricular infusion every 2 weeks; first ERT for CNS disease |
Substrate Reduction Therapy (SRT)
| Drug | Disease | Mechanism |
|---|---|---|
| Miglustat (Zavesca) | Gaucher type I (mild-moderate), Niemann-Pick C | Inhibits glucosylceramide synthase; oral; also stabilizes NPC1 protein trafficking |
| Eliglustat (Cerdelga) | Gaucher type I | Oral glucosylceramide synthase inhibitor; requires CYP2D6 genotyping before use (poor metabolizers: lower dose; ultra-rapid: not recommended) |
Gene Therapies (Approved)
| Drug | Disease | Vector/Mechanism | Details |
|---|---|---|---|
| Onasemnogene abeparvovec (Zolgensma) | SMA (type I, <2 years) | AAV9 delivering SMN1 | Single IV infusion; one of the most expensive therapies (>$2M) |
| Valoctocogene roxaparvovec (Roctavian) | Hemophilia A | AAV5 delivering FVIII gene | Single IV infusion; reduces/eliminates factor infusions |
| Etranacogene dezaparvovec (Hemgenix) | Hemophilia B | AAV5 delivering FIX Padua gain-of-function variant | Single IV infusion; near-normal FIX levels achieved |
| Delandistrogene moxeparvovec (Elevidys) | DMD (ages 4–5) | AAVrh74 delivering micro-dystrophin | Single IV infusion; accelerated approval |
| Exagamglogene autotemcel (Casgevy) | SCD, β-thalassemia | CRISPR/Cas9 ex vivo editing of BCL11A enhancer in HSCs | First CRISPR-based therapy approved; reactivates fetal hemoglobin |
| Lovotibeglogene autotemcel (Lyfgenia) | SCD | Lentiviral vector delivering anti-sickling β-globin to HSCs | Ex vivo gene addition to autologous HSCs |
| Betibeglogene autotemcel (Zynteglo) | β-Thalassemia | Lentiviral vector delivering functional β-globin | Ex vivo; for transfusion-dependent β-thal |
| Voretigene neparvovec (Luxturna) | RPE65-mediated retinal dystrophy | AAV2 delivering RPE65 | Subretinal injection; for biallelic RPE65 mutations causing Leber congenital amaurosis or retinitis pigmentosa |
Molecular Modulators & Targeted Therapies
| Drug | Disease | Mechanism |
|---|---|---|
| Elexacaftor/tezacaftor/ivacaftor (Trikafta) | CF (at least one F508del) | CFTR correctors + potentiator |
| Nusinersen (Spinraza) | SMA | Antisense oligonucleotide; modifies SMN2 splicing to increase full-length SMN protein |
| Risdiplam (Evrysdi) | SMA | Oral small molecule SMN2 splicing modifier |
| Migalastat (Galafold) | Fabry disease (amenable GLA mutations) | Pharmacologic chaperone; stabilizes misfolded α-galactosidase A |
| Vosoritide (Voxzogo) | Achondroplasia (children ≥5) | C-type natriuretic peptide analog; counteracts FGFR3 signaling to increase linear growth |
| Selumetinib (Koselugo) | NF1 plexiform neurofibromas | MEK1/2 inhibitor |
| Everolimus (Afinitor) | TSC (SEGA, AML, LAM) | mTOR inhibitor |
| Sapropterin (Kuvan) | PKU (BH4-responsive) | Synthetic BH4 cofactor for PAH |
| Pegvaliase (Palynziq) | PKU (adults) | PEGylated phenylalanine ammonia lyase; enzyme substitution |
| Nitisinone (Orfadin) | Tyrosinemia type I | Inhibits 4-hydroxyphenylpyruvate dioxygenase (upstream block prevents toxic metabolite formation) |
| Trofinetide (Daybue) | Rett syndrome | Synthetic analog of glycine-proline-glutamate; IGF-1 tripeptide analog |
| Belzutifan (Welireg) | VHL-associated tumors | HIF-2α inhibitor |
38 Abbreviations Master List & Reference Tables
Abbreviations
| Abbreviation | Full Term |
|---|---|
| ACMG | American College of Medical Genetics and Genomics |
| AD | Autosomal dominant |
| AFLP | Acute fatty liver of pregnancy |
| AMP | Association for Molecular Pathology |
| AR | Autosomal recessive |
| ASD | Autism spectrum disorder (or atrial septal defect, context-dependent) |
| AVSD | Atrioventricular septal defect |
| BWS | Beckwith-Wiedemann syndrome |
| CBAVD | Congenital bilateral absence of the vas deferens |
| cfDNA | Cell-free DNA |
| CGH | Comparative genomic hybridization |
| CK | Creatine kinase |
| CMA | Chromosomal microarray |
| CNV | Copy number variant |
| CPIC | Clinical Pharmacogenetics Implementation Consortium |
| CRRT | Continuous renal replacement therapy |
| CVS | Chorionic villus sampling |
| DMD | Duchenne muscular dystrophy (or dystrophin gene) |
| EDS | Ehlers-Danlos syndrome |
| EMA | Eosin-5-maleimide (binding test) |
| ERT | Enzyme replacement therapy |
| FAO | Fatty acid oxidation |
| FAP | Familial adenomatous polyposis |
| FH | Familial hypercholesterolemia |
| FISH | Fluorescence in situ hybridization |
| GINA | Genetic Information Nondiscrimination Act |
| GSD | Glycogen storage disease |
| GWAS | Genome-wide association study |
| HELLP | Hemolysis, Elevated Liver enzymes, Low Platelets |
| HNPCC | Hereditary nonpolyposis colorectal cancer |
| HSCT | Hematopoietic stem cell transplant |
| IEM | Inborn error of metabolism |
| IHC | Immunohistochemistry |
| ISCN | International System for Human Cytogenomic Nomenclature |
| IUGR | Intrauterine growth restriction |
| LOH | Loss of heterozygosity |
| MCADD | Medium-chain acyl-CoA dehydrogenase deficiency |
| MELAS | Mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes |
| MEN | Multiple endocrine neoplasia |
| MERRF | Myoclonic epilepsy with ragged red fibers |
| MLPA | Multiplex ligation-dependent probe amplification |
| MMR | DNA mismatch repair |
| MPS | Mucopolysaccharidosis |
| MSI | Microsatellite instability |
| mtDNA | Mitochondrial DNA |
| NBS | Newborn screening |
| NGS | Next-generation sequencing |
| NIPT | Non-invasive prenatal testing |
| NSGC | National Society of Genetic Counselors |
| NT | Nuchal translucency |
| OTC | Ornithine transcarbamylase |
| PARP | Poly(ADP-ribose) polymerase |
| PGT | Preimplantation genetic testing |
| PKU | Phenylketonuria |
| PPV | Positive predictive value |
| RUSP | Recommended Uniform Screening Panel |
| SCD | Sickle cell disease |
| SMA | Spinal muscular atrophy |
| SNP | Single nucleotide polymorphism |
| SNV | Single nucleotide variant |
| SRT | Substrate reduction therapy |
| TCD | Transcranial Doppler |
| TSC | Tuberous sclerosis complex |
| UPD | Uniparental disomy |
| VHL | Von Hippel-Lindau |
| VLCFA | Very long-chain fatty acids |
| VOUS/VUS | Variant of uncertain significance |
| WES | Whole exome sequencing |
| WGS | Whole genome sequencing |
| XLD | X-linked dominant |
| XLR | X-linked recessive |
Inheritance Pattern Quick Reference
| Pattern | Key Pedigree Features | Recurrence Risk | Classic Examples |
|---|---|---|---|
| AD | Vertical transmission; male-to-male possible; variable expressivity | 50% per offspring | Marfan, NF1, Huntington, achondroplasia, ADPKD, FH |
| AR | Horizontal pattern; consanguinity increases risk; parents unaffected carriers | 25% per offspring (carrier parents) | CF, SCD, PKU, Tay-Sachs, SMA, Wilson, hemochromatosis |
| XLR | Males affected; no male-to-male transmission; carrier females | 50% sons affected, 50% daughters carriers (from carrier mother) | DMD/BMD, hemophilia A/B, G6PD, Fabry, Hunter (MPS II) |
| XLD | Both sexes affected; often lethal in males; affected fathers transmit to all daughters | 50% all offspring; 100% daughters of affected males | Rett, incontinentia pigmenti, Aicardi, X-linked hypophosphatemic rickets |
| Mitochondrial | Maternal transmission; variable expressivity (heteroplasmy) | All children of affected mother at risk; affected father cannot transmit | MELAS, MERRF, LHON |
| Multifactorial | Familial clustering without clear Mendelian pattern | Empiric (2–5% first-degree relatives) | NTDs, cleft lip, CHD, T2DM, schizophrenia |
Common Trinucleotide Repeat Disorders
| Disorder | Gene | Repeat | Normal | Pathogenic | Anticipation / Transmission Bias |
|---|---|---|---|---|---|
| Huntington disease | HTT | CAG | ≤26 | ≥40 | Paternal (expansion in spermatogenesis) |
| Fragile X syndrome | FMR1 | CGG | 5–44 | >200 | Maternal (expansion from premutation → full mutation only maternally) |
| Myotonic dystrophy 1 | DMPK | CTG | 5–34 | >50 | Maternal (congenital form with massive expansion) |
| Friedreich ataxia | FXN | GAA | 5–33 | ≥66 | AR; no true anticipation; loss of function (reduced frataxin) |
| Spinocerebellar ataxias (multiple) | Various | CAG | Variable | Variable | Paternal in most SCAs |
| Spinal-bulbar muscular atrophy (Kennedy) | AR (androgen receptor) | CAG | 9–36 | ≥38 | X-linked; mild anticipation |