Neurology
Every diagnosis, classification, medication, diagnostic modality, examination technique, and management strategy — from stroke to seizure, demyelinating disease to neuromuscular disorders — in one place.
01 Neuroanatomy — Brain & Cranial Nerves
Cerebral Cortex — Lobes & Functions
The frontal lobe occupies the anterior third of each hemisphere, bounded posteriorly by the central sulcus and inferiorly by the lateral (Sylvian) fissure. It contains the primary motor cortex (precentral gyrus — Brodmann area 4), the premotor cortex (area 6), and the prefrontal cortex (executive function, judgment, personality, working memory). Broca's area (areas 44–45) in the dominant (usually left) inferior frontal gyrus controls expressive language — damage produces non-fluent (Broca's) aphasia: halting, effortful speech with preserved comprehension.
The parietal lobe lies posterior to the central sulcus and above the lateral fissure. Its primary somatosensory cortex (postcentral gyrus — areas 3, 1, 2) processes contralateral tactile, proprioceptive, and pain input. The inferior parietal lobule in the dominant hemisphere integrates language processing (angular gyrus — area 39 — alexia with agraphia when damaged). Non-dominant parietal lesions produce hemispatial neglect — the patient ignores the contralateral side of space.
The temporal lobe lies below the lateral fissure. It houses the primary auditory cortex (Heschl's gyrus — areas 41, 42), the hippocampus (medial temporal — memory formation), and Wernicke's area (posterior superior temporal gyrus — area 22) in the dominant hemisphere — damage produces fluent (Wernicke's) aphasia: flowing but meaningless speech with impaired comprehension. The occipital lobe is the most posterior, containing the primary visual cortex (area 17 along the calcarine fissure). Unilateral occipital lesions produce contralateral homonymous hemianopia.
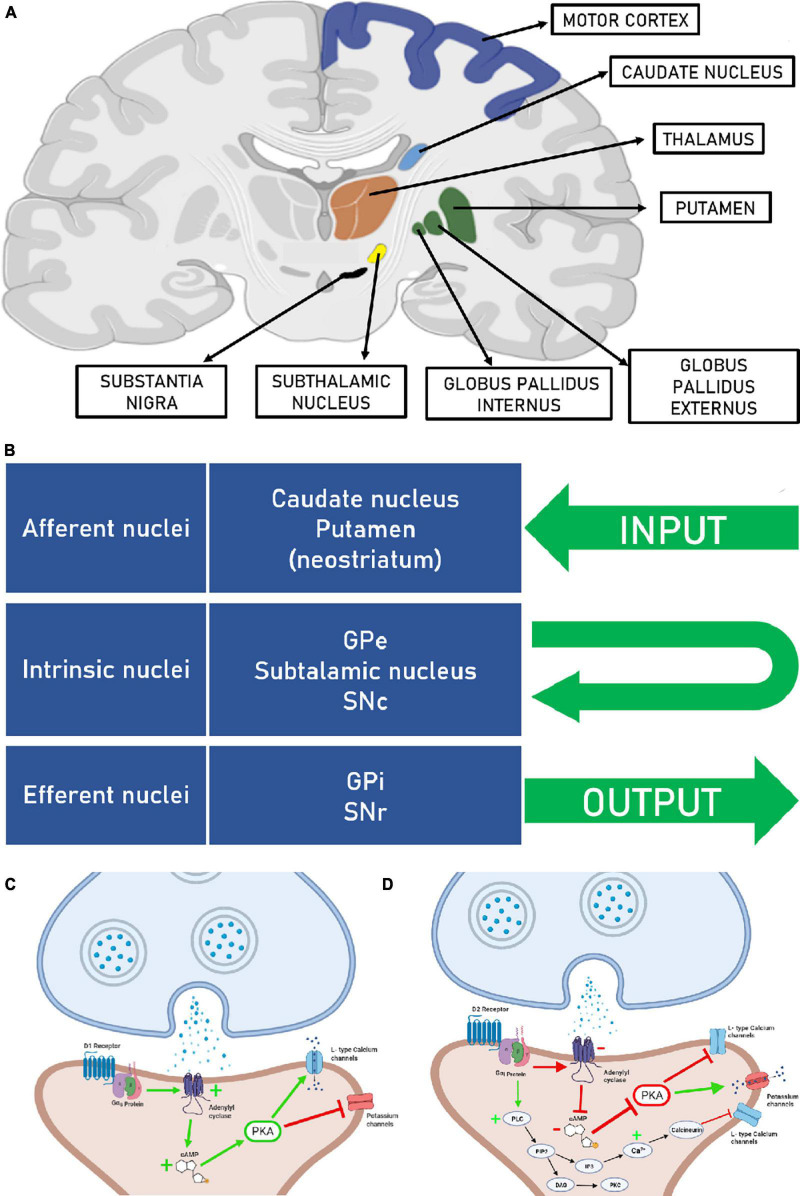
Subcortical Structures
The basal ganglia (caudate nucleus, putamen, globus pallidus, subthalamic nucleus, substantia nigra) regulate motor planning, initiation, and inhibition. The caudate + putamen = striatum (primary input nuclei, receiving cortical glutamatergic input); the putamen + globus pallidus = lentiform nucleus. Degeneration of dopaminergic neurons in the substantia nigra pars compacta produces Parkinson's disease. The thalamus is the relay station for nearly all sensory and motor pathways to the cortex. The hypothalamus controls autonomic function, thermoregulation, hunger, thirst, circadian rhythms, and hormonal release via the pituitary. The limbic system (hippocampus, amygdala, cingulate gyrus) mediates memory encoding, emotion, and motivation.
Brainstem
The brainstem connects the cerebrum to the spinal cord and contains cranial nerve nuclei, ascending/descending tracts, and the reticular activating system (consciousness). Three divisions from rostral to caudal: midbrain (CN III, IV; cerebral peduncles; superior/inferior colliculi; substantia nigra; red nucleus), pons (CN V, VI, VII, VIII; pontine nuclei; middle cerebellar peduncles), and medulla oblongata (CN IX, X, XI, XII; pyramids where the corticospinal tracts decussate; nucleus solitarius for visceral sensation; dorsal motor nucleus of vagus). The medulla contains the vital respiratory and cardiovascular centers — compression causes bradycardia, hypertension, and irregular respirations (Cushing's triad).
Cranial Nerves
| CN | Name | Type | Function | Key Lesion Sign |
|---|---|---|---|---|
| I | Olfactory | Sensory | Smell | Anosmia (frontal lobe tumors, trauma) |
| II | Optic | Sensory | Vision, pupillary light reflex (afferent) | Visual field defects, APD (Marcus Gunn pupil) |
| III | Oculomotor | Motor | SR, IR, MR, IO; lid elevation; parasympathetic pupil constriction | "Down and out" eye, ptosis, mydriasis |
| IV | Trochlear | Motor | Superior oblique (intorts and depresses) | Vertical diplopia worse on downgaze; head tilt |
| V | Trigeminal | Both | Facial sensation (V1-V2-V3); muscles of mastication | Facial numbness, jaw deviation toward lesion, absent corneal reflex (afferent) |
| VI | Abducens | Motor | Lateral rectus (abduction) | Medial eye deviation, horizontal diplopia; false localizing sign in raised ICP |
| VII | Facial | Both | Facial expression muscles; taste anterior 2/3 tongue; lacrimation, salivation | UMN: contralateral lower face weakness; LMN (Bell's): entire ipsilateral face |
| VIII | Vestibulocochlear | Sensory | Hearing (cochlear), balance (vestibular) | Sensorineural hearing loss, vertigo, nystagmus |
| IX | Glossopharyngeal | Both | Taste posterior 1/3 tongue; pharyngeal sensation; carotid body/sinus | Absent gag reflex (afferent limb), glossopharyngeal neuralgia |
| X | Vagus | Both | Palatal/pharyngeal/laryngeal motor; parasympathetic to thoracoabdominal viscera | Hoarseness (recurrent laryngeal), uvula deviates away from lesion, dysphagia |
| XI | Accessory | Motor | Sternocleidomastoid, trapezius | Weakness of head turning (SCM), shoulder shrug (trapezius) |
| XII | Hypoglossal | Motor | Tongue movement | Tongue deviates toward the lesion (LMN); away from lesion in UMN |
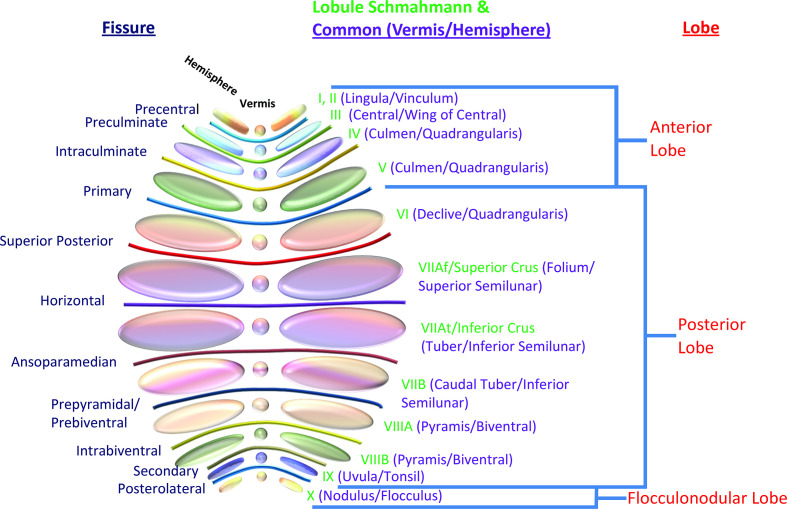
Cerebellum
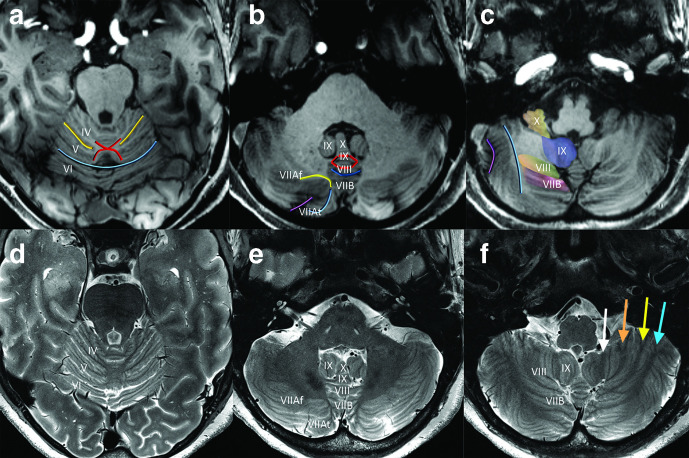
The cerebellum coordinates voluntary movement, balance, and motor learning. Three functional divisions: vestibulocerebellum (flocculonodular lobe — balance, eye movements; lesions cause truncal ataxia and nystagmus), spinocerebellum (vermis + paravermal zones — posture, gait, limb coordination; vermis lesions cause wide-based gait ataxia), and cerebrocerebellum (lateral hemispheres — motor planning, timing; lesions cause ipsilateral limb dysmetria, intention tremor, dysdiadochokinesia). Cerebellar signs are always ipsilateral to the lesion — unlike cerebral hemisphere lesions, which produce contralateral deficits.
02 Neuroanatomy — Spinal Cord & Vascular Territories
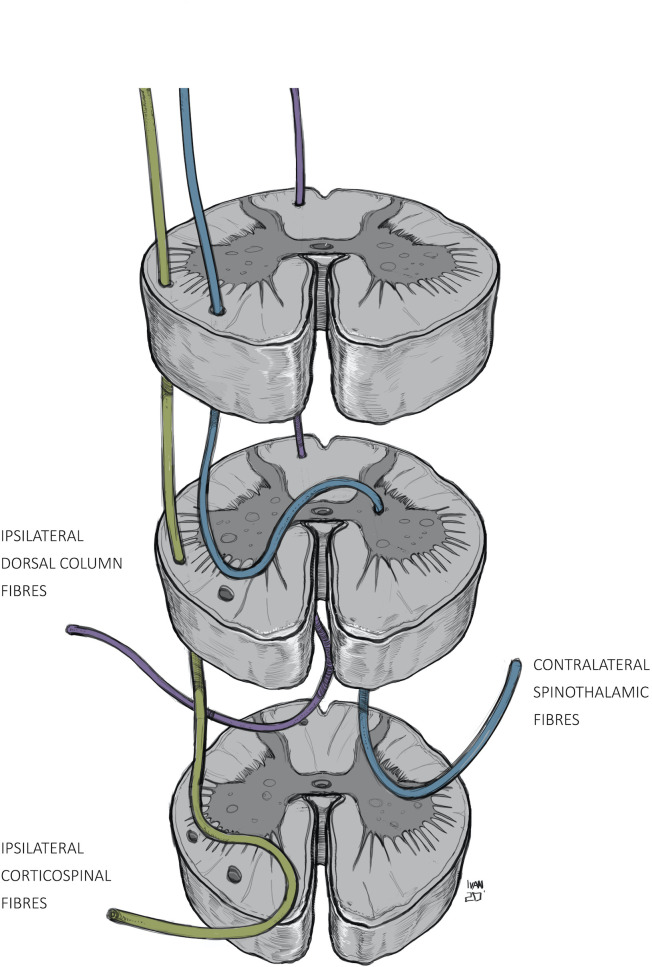
Spinal Cord Tracts
| Tract | Location in Cord | Function | Decussation | Lesion Deficit |
|---|---|---|---|---|
| Corticospinal (lateral) | Lateral column | Voluntary motor — contralateral limbs | Pyramidal decussation (caudal medulla) | Ipsilateral UMN weakness below lesion |
| Dorsal columns (fasciculus gracilis/cuneatus) | Posterior column | Fine touch, vibration, proprioception | Nucleus gracilis/cuneatus (medulla) → medial lemniscus | Ipsilateral loss of vibration, proprioception below lesion |
| Spinothalamic (lateral) | Anterolateral column | Pain, temperature | At spinal cord level (anterior white commissure — 1-2 levels above entry) | Contralateral loss of pain/temperature below lesion |
| Anterior corticospinal | Anterior column | Axial/proximal motor (bilateral) | At level of destination | Minimal deficit (bilateral innervation) |
| Spinocerebellar (dorsal/ventral) | Lateral column | Unconscious proprioception to cerebellum | Dorsal: ipsilateral; Ventral: double-cross | Ipsilateral limb ataxia |
Spinal Cord Syndromes
| Syndrome | Mechanism | Pattern | Prognosis |
|---|---|---|---|
| Complete transection | Trauma, compression | Loss of all motor/sensory below level; spinal shock initially (flaccid, areflexic), then UMN pattern | Poor recovery below level |
| Brown-Séquard (hemisection) | Penetrating trauma, tumor | Ipsilateral: motor loss + dorsal column loss; Contralateral: spinothalamic loss (pain/temp 1-2 levels below) | Best prognosis of incomplete syndromes |
| Central cord | Hyperextension in cervical spondylosis | Upper extremity weakness > lower (hands worst), variable sensory loss, bladder dysfunction | LE recovers first, then bladder, then UE; hands last |
| Anterior cord | Anterior spinal artery occlusion | Motor paralysis + loss of pain/temp bilaterally; preserved dorsal column (proprioception, vibration) | Poor — worst prognosis of incomplete syndromes |
| Posterior cord | B12 deficiency, tabes dorsalis, MS | Loss of proprioception, vibration bilaterally; preserved motor and pain/temp | Variable — depends on cause |
| Conus medullaris | Tumor, disc at L1-L2 | Early bowel/bladder dysfunction (areflexic bladder), saddle anesthesia, symmetric, mild leg weakness | Often irreversible bladder dysfunction |
| Cauda equina | Large disc herniation, tumor | Asymmetric radicular pain, LMN weakness, saddle anesthesia, urinary retention (late) | Surgical emergency — better if decompressed < 48 hours |
Cerebral Vascular Territories
| Artery | Territory | Stroke Syndrome |
|---|---|---|
| Anterior cerebral artery (ACA) | Medial frontal/parietal cortex, supplementary motor area | Contralateral leg weakness > arm, abulia, urinary incontinence, alien hand (if corpus callosum involved) |
| Middle cerebral artery (MCA) | Lateral frontal, parietal, temporal cortex; basal ganglia (lenticulostriate branches) | Contralateral face/arm weakness > leg, hemianesthesia, homonymous hemianopia; dominant: aphasia; non-dominant: neglect |
| Posterior cerebral artery (PCA) | Occipital lobe, medial temporal, thalamus | Contralateral homonymous hemianopia with macular sparing, alexia without agraphia (dominant), memory deficits (hippocampus) |
| Basilar artery | Pons, midbrain, cerebellum | Locked-in syndrome (ventral pons); coma; cranial nerve palsies + long tract signs |
| PICA (posterior inferior cerebellar) | Lateral medulla, inferior cerebellum | Lateral medullary (Wallenberg) syndrome: ipsilateral facial pain/temp loss, Horner's, ataxia, dysphagia; contralateral body pain/temp loss |
| AICA (anterior inferior cerebellar) | Lateral pons, anterior inferior cerebellum | Ipsilateral facial paralysis, hearing loss, vertigo, ataxia; contralateral body pain/temp loss |
| SCA (superior cerebellar) | Superior cerebellum, dorsal midbrain | Ipsilateral ataxia, contralateral pain/temp loss, ipsilateral Horner's |
| Anterior spinal artery | Anterior 2/3 of spinal cord | Anterior cord syndrome (bilateral motor/pain-temp loss, preserved proprioception) |
03 The Neurological Examination
Mental Status
Assess level of consciousness (alert, lethargic, obtunded, stuporous, comatose), orientation (person, place, time, situation), attention (serial 7s, spell "WORLD" backwards, digit span — normal forward is 7 ± 2), language (fluency, comprehension, repetition, naming, reading, writing — localizes aphasia type), memory (immediate recall, short-term at 5 minutes, long-term), and executive function (Luria hand sequences, clock drawing, verbal fluency — name as many animals as possible in 60 seconds; normal ≥ 12). The Mini-Mental State Examination (MMSE) scores 0–30 (normal ≥ 24); the Montreal Cognitive Assessment (MoCA) is more sensitive for mild cognitive impairment (normal ≥ 26/30).
Cranial Nerve Examination
CN II: Visual acuity (Snellen chart), visual fields by confrontation (each eye separately — wiggle finger in each quadrant), fundoscopy (papilledema, optic disc pallor), pupillary light reflex (direct and consensual), swinging flashlight test for relative afferent pupillary defect (APD/Marcus Gunn pupil — pupil dilates when light swings to it, indicating optic nerve damage on that side). CN III/IV/VI: Extraocular movements — trace an "H" pattern; assess for nystagmus, internuclear ophthalmoplegia (INO — failure of adduction on lateral gaze with nystagmus of abducting eye; caused by MLF lesion; bilateral INO strongly suggests MS in young patients). CN V: Sensation in V1, V2, V3 distributions; corneal reflex (afferent V1, efferent VII); jaw clench strength. CN VII: Forehead wrinkling, eye closure, smile, puff cheeks — distinguish UMN (forehead spared because of bilateral innervation) from LMN (entire ipsilateral face). CN VIII: Weber (tuning fork on vertex — lateralizes to conductive loss, away from sensorineural) and Rinne (compare air vs bone conduction). CN IX/X: Gag reflex, palate elevation ("say ahh" — uvula deviates away from weak side), voice quality. CN XI: Shoulder shrug (trapezius), head turn against resistance (SCM — turns head to opposite side). CN XII: Tongue protrusion (deviates toward lesion in LMN), fasciculations.
Motor Examination
Assess bulk (atrophy — LMN or disuse), tone (spasticity = UMN, velocity-dependent "clasp-knife"; rigidity = basal ganglia, velocity-independent "lead-pipe" or "cogwheel" with superimposed tremor; flaccidity = LMN or acute UMN/spinal shock), and strength using the MRC scale:
| Grade | Description |
|---|---|
| 5 | Full strength against full resistance |
| 4 | Movement against gravity and some resistance |
| 3 | Movement against gravity but not resistance |
| 2 | Movement with gravity eliminated |
| 1 | Visible/palpable muscle contraction but no movement |
| 0 | No contraction |
Test proximal and distal muscle groups in all four extremities. Key patterns: UMN — weakness of extensors in upper extremity (arm extensors, finger extensors, wrist extensors) and flexors in lower extremity (hip flexors, knee flexors, ankle dorsiflexors), giving the classic "pyramidal" pattern. LMN — weakness in specific myotomal or nerve distribution with atrophy and fasciculations.
Sensory Examination
Test light touch (cotton wisp — dorsal columns primarily), pinprick (spinothalamic), temperature (cold tuning fork — spinothalamic), vibration (128 Hz tuning fork on bony prominences — dorsal columns; compare distal to proximal), proprioception (joint position sense at great toe — dorsal columns), and cortical sensation (stereognosis, graphesthesia, two-point discrimination — parietal lobe). Dermatomal patterns suggest radiculopathy; stocking-glove pattern suggests peripheral neuropathy; a sensory level suggests myelopathy.
Reflexes
| Reflex | Root Level | Normal Response |
|---|---|---|
| Biceps | C5-C6 | Elbow flexion |
| Brachioradialis | C5-C6 | Elbow flexion, forearm pronation |
| Triceps | C7-C8 | Elbow extension |
| Patellar (knee jerk) | L3-L4 | Knee extension |
| Achilles (ankle jerk) | S1-S2 | Plantar flexion |
Grade: 0 = absent, 1+ = hypoactive, 2+ = normal, 3+ = hyperactive without clonus, 4+ = hyperactive with clonus. Hyperreflexia = UMN; hyporeflexia/areflexia = LMN. Babinski sign (upgoing plantar response) = UMN lesion (normal in infants < 1 year). Hoffman's sign (flicking the distal phalanx of the middle finger causes thumb/index flexion) = cervical myelopathy/UMN. Clonus (≥ 3 beats sustained) = UMN.
Coordination & Gait
Finger-to-nose and heel-to-shin test cerebellar function (dysmetria = overshoot/undershoot). Rapid alternating movements (dysdiadochokinesia = cerebellar). Romberg test: patient stands feet together, eyes closed — positive (falls) = proprioceptive or vestibular dysfunction (not cerebellar — cerebellar patients are unstable even with eyes open). Gait patterns: spastic (stiff, circumduction — UMN), steppage (foot drop, high knee — peroneal nerve/L5), waddling (hip girdle myopathy), ataxic/wide-based (cerebellar), magnetic/apraxic (frontal lobe, NPH), festinating (small shuffling steps, forward lean — Parkinson's).
04 Key Terminology
| Term | Definition |
|---|---|
| Aphasia | Impaired language (Broca's = non-fluent expressive; Wernicke's = fluent receptive; Global = both; Conduction = impaired repetition with preserved fluency/comprehension) |
| Dysarthria | Impaired speech articulation (motor, not language) — cerebellar, LMN, UMN, or extrapyramidal |
| Ataxia | Impaired coordination — cerebellar (appendicular or truncal), sensory (proprioceptive loss), or vestibular |
| Nystagmus | Involuntary rhythmic eye oscillation — named by fast phase direction; peripheral (unidirectional, inhibited by fixation, with vertigo) vs central (multidirectional, not inhibited, may lack vertigo) |
| Myoclonus | Brief, shock-like involuntary jerks (cortical, subcortical, spinal, or peripheral origin) |
| Fasciculation | Spontaneous firing of a motor unit visible under skin — LMN sign (denervation); benign fasciculations also exist |
| Paresis vs Plegia | Paresis = weakness; plegia = complete paralysis (hemiparesis vs hemiplegia, paraparesis vs paraplegia, tetraparesis vs tetraplegia) |
| Radiculopathy | Nerve root dysfunction producing pain, sensory loss, weakness, reflex changes in a dermatomal/myotomal pattern |
| Myelopathy | Spinal cord dysfunction — UMN signs below the level, sensory level, bowel/bladder involvement |
| Plexopathy | Brachial or lumbosacral plexus dysfunction — multiple nerve/root distributions affected simultaneously |
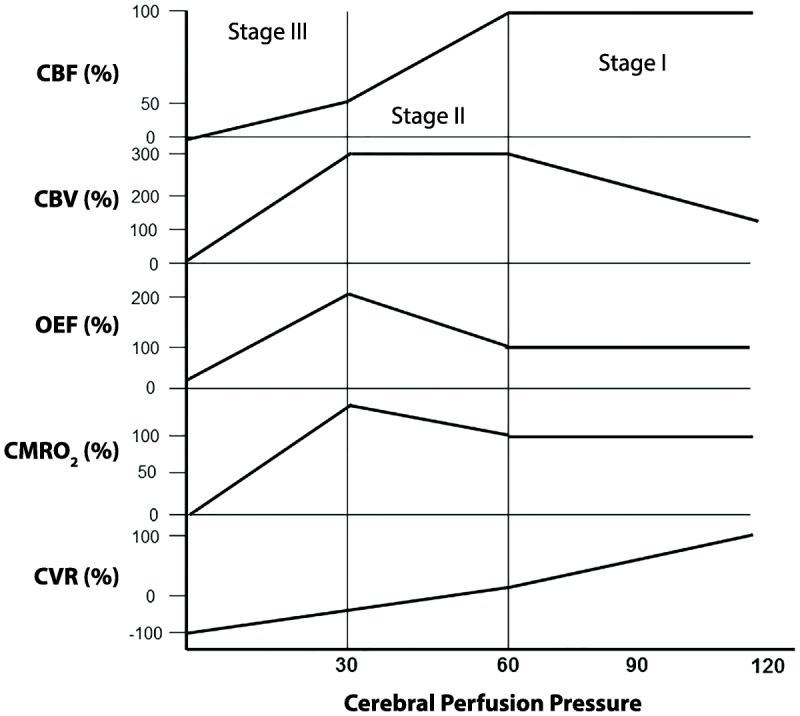
05 Acute Ischemic Stroke Cerebrovascular
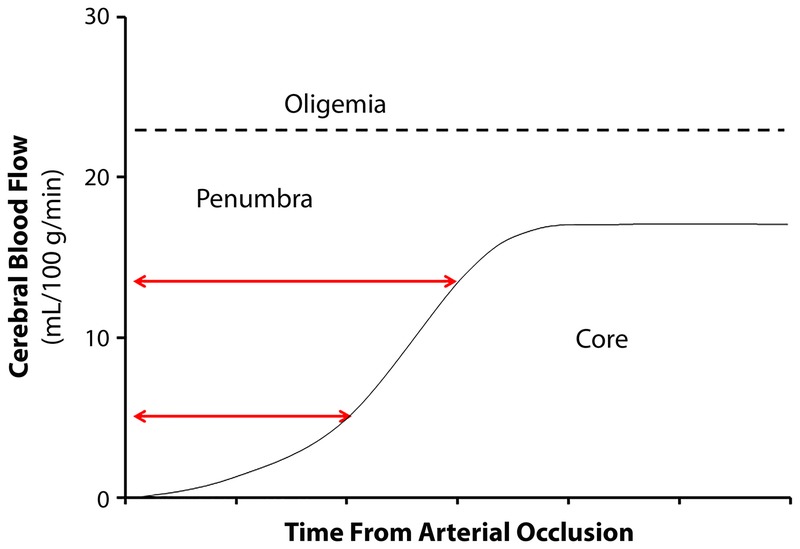
Ischemic stroke accounts for ~87% of all strokes. Sudden occlusion of a cerebral artery causes ischemia in the downstream territory. The ischemic penumbra — tissue that is hypoperfused but not yet infarcted — is the target of acute reperfusion therapy. Every minute of untreated large-vessel occlusion destroys ~1.9 million neurons (Saver, 2006). "Time is brain" is literal, not metaphorical.
TOAST Classification — Stroke Etiology
| Category | Mechanism | Key Features |
|---|---|---|
| Large-artery atherosclerosis | Atherothrombotic stenosis/occlusion of major cervical or intracranial artery | ≥ 50% stenosis or occlusion; cortical or large subcortical infarct (> 1.5 cm); carotid duplex/CTA/MRA confirms |
| Cardioembolism | Embolism from cardiac source | Atrial fibrillation, valvular disease, recent MI, dilated cardiomyopathy, patent foramen ovale, endocarditis; often involves multiple territories |
| Small-vessel occlusion (lacunar) | Lipohyalinosis of penetrating arterioles | Subcortical infarcts < 1.5 cm; classic lacunar syndromes (pure motor, pure sensory, ataxic hemiparesis, dysarthria-clumsy hand, mixed sensorimotor) |
| Other determined etiology | Dissection, vasculitis, hypercoagulable state, sickle cell, moyamoya, etc. | Young patients, no conventional risk factors, specific lab/imaging evidence |
| Cryptogenic / ESUS | No cause identified despite thorough workup | ~25% of ischemic strokes; many ultimately found to have occult paroxysmal AF or PFO |
NIH Stroke Scale (NIHSS)
The NIHSS is the standardized tool for quantifying stroke severity (0–42 points). It guides treatment decisions: NIHSS ≥ 6 strongly suggests a large vessel occlusion warranting emergent CTA and potential thrombectomy. Key items:
| Item | Tests | Score Range |
|---|---|---|
| 1a. Level of consciousness | Alertness | 0–3 |
| 1b. LOC questions | Month, age | 0–2 |
| 1c. LOC commands | Open/close eyes, grip/release | 0–2 |
| 2. Best gaze | Horizontal eye movements | 0–2 |
| 3. Visual fields | Confrontation | 0–3 |
| 4. Facial palsy | Show teeth, raise eyebrows | 0–3 |
| 5. Motor — arms | Hold arms at 90° (sitting) or 45° (supine) for 10 sec | 0–4 each |
| 6. Motor — legs | Hold legs at 30° supine for 5 sec | 0–4 each |
| 7. Limb ataxia | Finger-nose, heel-shin | 0–2 |
| 8. Sensory | Pinprick | 0–2 |
| 9. Best language | Naming, reading, describing | 0–3 |
| 10. Dysarthria | Read words/sentences | 0–2 |
| 11. Extinction/inattention | Double simultaneous stimulation | 0–2 |
Acute Management — IV Thrombolysis
Alteplase (tPA) 0.9 mg/kg IV (max 90 mg), 10% as bolus, remainder over 60 minutes. Window: within 4.5 hours of symptom onset (or last known well). The NINDS trial (1995) established the 3-hour window; the ECASS III (2008) extended to 4.5 hours with additional exclusion criteria (age > 80, NIHSS > 25, oral anticoagulant use, history of both stroke and diabetes). Tenecteplase (0.25 mg/kg IV single bolus, max 25 mg) is increasingly used as an alternative — simpler administration, and the EXTEND-IA TNK trial showed non-inferiority to alteplase for LVO with better early reperfusion. Key contraindications: SBP > 185/110 despite treatment, platelets < 100K, INR > 1.7, recent major surgery/trauma (< 14 days), active internal bleeding, intracranial hemorrhage on CT.
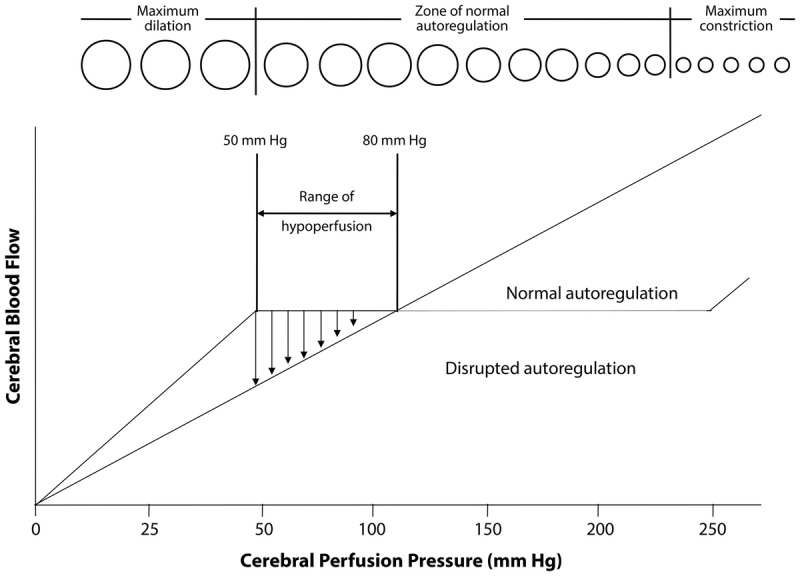
Pre-tPA: must lower to < 185/110 before administering (labetalol 10–20 mg IV, nicardipine drip 5 mg/hr titrated). Post-tPA: maintain < 180/105 for 24 hours. No tPA given: permissive hypertension up to 220/120 (autoregulatory response to maintain penumbral perfusion) — treat only if > 220/120 or end-organ damage. Rapid BP reduction in acute ischemic stroke without reperfusion therapy worsens outcomes.
Mechanical Thrombectomy
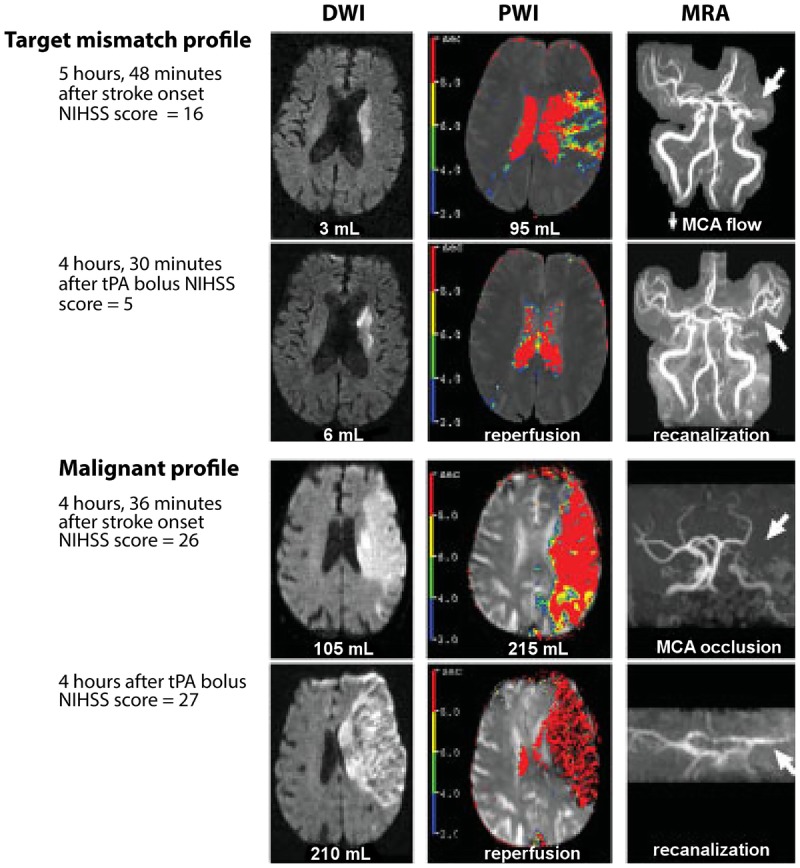
For large vessel occlusion (LVO) — ICA, M1 MCA, and in select cases M2 MCA or basilar artery. Five landmark 2015 trials (MR CLEAN, ESCAPE, EXTEND-IA, SWIFT PRIME, REVASCAT) demonstrated overwhelming benefit of endovascular thrombectomy + IV tPA vs tPA alone. Standard window: within 6 hours. Extended window to 24 hours for selected patients with favorable perfusion imaging (core infarct < 70 mL on CTP or DWI, and significant mismatch between core and penumbra) — established by DAWN (2018) and DEFUSE 3 (2018). NNT for good functional outcome (mRS 0–2) is approximately 2.6 — one of the most effective interventions in all of medicine.
Secondary Prevention
Antiplatelet: ASA 81–325 mg daily for non-cardioembolic stroke; dual antiplatelet (ASA + clopidogrel) for 21 days after minor stroke/TIA (NIHSS ≤ 3), then single agent (POINT trial). Cardioembolic (AF): anticoagulation with DOAC (apixaban, rivaroxaban, dabigatran, edoxaban) — superior to warfarin for non-valvular AF; start timing depends on infarct size (small infarct: 1–3 days; medium: 6–8 days; large/hemorrhagic transformation: 12–14 days — the "1-3-6-12" rule). Statins: high-intensity (atorvastatin 80 mg) for all atherosclerotic strokes. Carotid stenosis ≥ 70% symptomatic: CEA or CAS within 2 weeks of event.
06 Hemorrhagic Stroke — ICH & SAH Cerebrovascular
Intracerebral Hemorrhage (ICH)
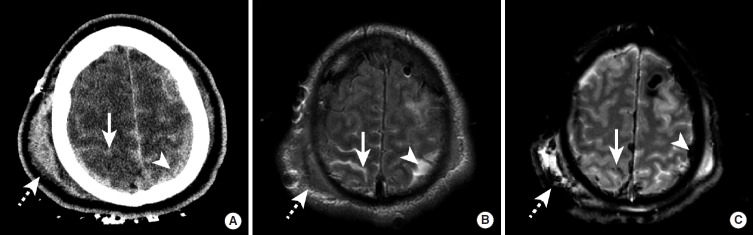
ICH accounts for ~13% of strokes but ~40% of stroke mortality. The most common cause is hypertensive vasculopathy — chronic hypertension weakens penetrating arterioles, forming Charcot-Bouchard microaneurysms that rupture. Typical locations: basal ganglia (putamen — most common, ~35%), thalamus (~20%), pons (~5%), cerebellum (~10%), lobar (~30%). Lobar ICH in elderly patients suggests cerebral amyloid angiopathy (CAA) — amyloid-beta deposits in cortical/leptomeningeal vessel walls causing recurrent lobar hemorrhages.
ICH Score (prognostic — mortality at 30 days):
| Component | Points |
|---|---|
| GCS 3–4 | 2 |
| GCS 5–12 | 1 |
| GCS 13–15 | 0 |
| ICH volume ≥ 30 mL | 1 |
| Intraventricular hemorrhage present | 1 |
| Infratentorial origin | 1 |
| Age ≥ 80 | 1 |
Score 0 = 0% mortality; 1 = 13%; 2 = 26%; 3 = 72%; 4 = 97%; 5 = 100%.
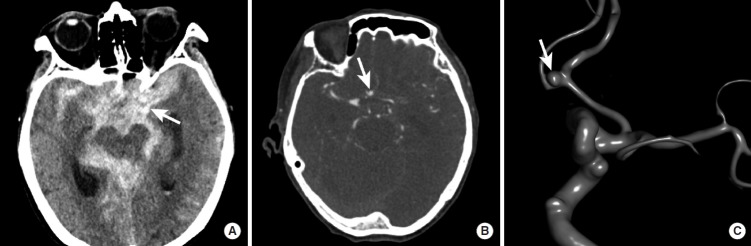
Management: Emergent BP reduction: SBP target < 140 mmHg within 1 hour (nicardipine drip preferred) — the INTERACT2 trial showed improved functional outcomes with intensive lowering. Reverse anticoagulation immediately: warfarin → PCC (Kcentra) 25–50 units/kg + vitamin K 10 mg IV; dabigatran → idarucizumab (Praxbind); rivaroxaban/apixaban → andexanet alfa or PCC. Platelet transfusion for antiplatelet-associated ICH is NOT recommended (PATCH trial showed harm). Surgical evacuation: consider for cerebellar ICH > 3 cm with deterioration, or supratentorial lobar ICH with GCS 9–12 and clot accessible (STICH II trial). External ventricular drain (EVD) for hydrocephalus from intraventricular hemorrhage.
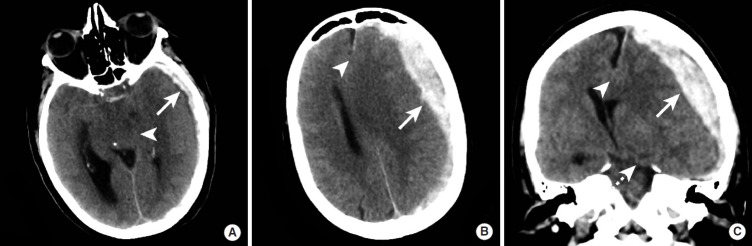
Subarachnoid Hemorrhage (SAH)
Nontraumatic SAH is caused by ruptured saccular (berry) aneurysm in ~85% of cases. The classic presentation is "worst headache of my life" — thunderclap onset peaking within seconds, often with nausea/vomiting, neck stiffness, photophobia, and loss of consciousness. Mortality is ~40% overall; 15% die before reaching the hospital. Most common aneurysm location: anterior communicating artery (AComm) (~30%), then posterior communicating artery (PComm) (~25%), MCA bifurcation (~20%).
Hunt-Hess Grade (Clinical Severity at Presentation)
| Grade | Clinical Description | Approximate Mortality |
|---|---|---|
| I | Asymptomatic or mild headache, slight nuchal rigidity | ~1% |
| II | Moderate-severe headache, nuchal rigidity, no focal deficit (except CN palsy) | ~5% |
| III | Drowsy/confused, mild focal deficit | ~19% |
| IV | Stupor, moderate-severe hemiparesis, early decerebrate posturing | ~42% |
| V | Deep coma, decerebrate posturing, moribund | ~77% |
Fisher Grade (CT Appearance — Predicts Vasospasm Risk)
| Grade | CT Finding | Vasospasm Risk |
|---|---|---|
| 1 | No blood detected | Low (21%) |
| 2 | Diffuse thin layer < 1 mm | Low (25%) |
| 3 | Localized clot ≥ 1 mm thick or diffuse > 1 mm | High (37%) |
| 4 | Diffuse or no SAH, but intraventricular/intracerebral hemorrhage present | Low (31%) |
SAH Management: (1) Secure the aneurysm within 24 hours — endovascular coiling (preferred for most locations, per ISAT trial) or surgical clipping. (2) Nimodipine 60 mg PO q4h × 21 days — the only proven agent for preventing delayed cerebral ischemia from vasospasm. (3) Euvolemia (avoid both hypo- and hypervolemia; "triple-H therapy" — hypertension, hypervolemia, hemodilution — is largely abandoned except for induced hypertension for symptomatic vasospasm). (4) Monitor for vasospasm (peak days 4–14): daily transcranial Doppler (MCA velocity > 200 cm/sec = severe spasm), neuro checks q1h. (5) EVD for hydrocephalus. (6) Seizure prophylaxis is controversial — levetiracetam if used; phenytoin is avoided (associated with worse cognitive outcomes).
Rebleeding: Highest risk in the first 24 hours (~4% in the first 24h, ~1–2%/day for the next 2 weeks). This is why early aneurysm securing is critical. Vasospasm / delayed cerebral ischemia (DCI): Days 4–14; peaks day 7–8; presents as new focal deficit or declining level of consciousness. Hydrocephalus: Acute (obstructive — within hours) or chronic communicating (weeks later — blood products impair CSF reabsorption at arachnoid granulations). Hyponatremia: Cerebral salt wasting (CSW — volume-depleted, treat with NS/hypertonic) vs SIADH (euvolemic, treat with fluid restriction) — distinguishing them is critical because fluid restriction in CSW worsens vasospasm. Seizures: ~5–10% of SAH patients.
07 Transient Ischemic Attack (TIA) Cerebrovascular
TIA is a transient episode of neurological dysfunction caused by focal brain, spinal cord, or retinal ischemia without acute infarction on imaging. The modern (tissue-based) definition does not use a 24-hour time cutoff — any episode with DWI-positive infarction is classified as stroke regardless of symptom duration. Most true TIAs last < 15 minutes. TIA is a medical emergency: the 90-day stroke risk after TIA is 10–15%, with half occurring within the first 48 hours.
ABCD2 Score — 2-Day Stroke Risk After TIA
| Factor | Criteria | Points |
|---|---|---|
| Age | ≥ 60 years | 1 |
| Blood pressure | SBP ≥ 140 or DBP ≥ 90 at presentation | 1 |
| Clinical features | Unilateral weakness = 2; speech disturbance without weakness = 1; other = 0 | 0–2 |
| Duration | ≥ 60 min = 2; 10–59 min = 1; < 10 min = 0 | 0–2 |
| Diabetes | Present | 1 |
Score 0–3: low risk (~1%); 4–5: moderate risk (~4%); 6–7: high risk (~8%). However, ABCD2 has limited sensitivity — current guidelines recommend emergent workup for all TIAs regardless of score. Workup: brain MRI with DWI, neurovascular imaging (CTA or MRA head/neck), echocardiography, cardiac monitoring (telemetry ≥ 24h, consider 30-day event monitor for cryptogenic), and labs (CBC, CMP, lipid panel, A1c, PT/INR).
08 Cerebral Venous Thrombosis Cerebrovascular
Thrombosis of the dural venous sinuses or cortical veins. Unlike arterial stroke, CVT often presents subacutely over days to weeks. Most common in young women (OCP use, pregnancy/postpartum, hormonal factors). Other risk factors: hypercoagulable states (Factor V Leiden, prothrombin G20210A, antiphospholipid syndrome), infections (sinusitis, mastoiditis, meningitis), dehydration, malignancy. Most commonly affected: superior sagittal sinus (~62%) and transverse/lateral sinus (~45%).
Presentation: Headache (>90% — often progressive, worse with Valsalva), papilledema, seizures (~40%), focal deficits (hemiparesis, aphasia), altered consciousness. Diagnosis: CT venography (CTV) or MR venography (MRV) — shows filling defect ("empty delta sign" on contrast CT is classic but insensitive). D-dimer may be elevated but a normal D-dimer does not exclude CVT. Treatment: Anticoagulation with heparin (UFH or LMWH) even if hemorrhagic infarction is present — counterintuitive but supported by evidence. Transition to warfarin (INR 2–3) or DOAC for 3–12 months depending on the underlying cause. Endovascular thrombectomy/thrombolysis reserved for deterioration despite anticoagulation.
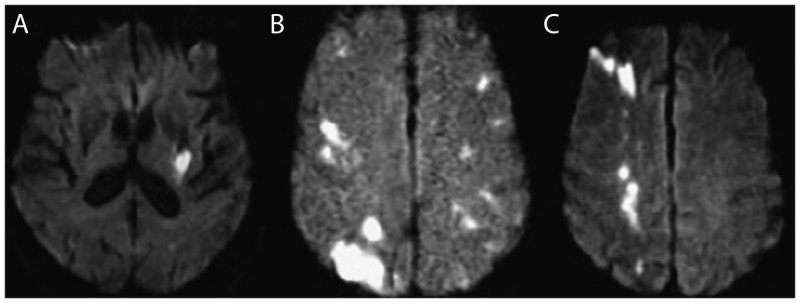
09 Seizure Classification (ILAE 2017) Epilepsy
The 2017 ILAE classification replaced the 1981 system. It organizes seizures by three features: (1) onset (focal, generalized, unknown), (2) awareness (aware vs impaired awareness for focal seizures), (3) motor vs non-motor manifestation.
Focal Onset Seizures
| Subtype | Awareness | Features |
|---|---|---|
| Focal aware (formerly "simple partial") | Preserved | Motor (clonic jerking, automatisms), sensory (tingling, visual phenomena), autonomic (flushing, nausea), psychic (déjà vu, fear) |
| Focal impaired awareness (formerly "complex partial") | Impaired | Staring, automatisms (lip smacking, hand fumbling), post-ictal confusion; most commonly from temporal lobe |
| Focal to bilateral tonic-clonic (formerly "secondary generalized") | Starts focal, then generalizes | Aura → loss of consciousness → bilateral convulsive activity |
Generalized Onset Seizures
| Type | Features | EEG Pattern |
|---|---|---|
| Tonic-clonic (grand mal) | Tonic phase (stiffening, 10–20 sec) → clonic phase (rhythmic jerking, 30–60 sec) → post-ictal confusion/sleepiness | Generalized polyspike-and-wave |
| Absence (petit mal) | Brief staring (5–30 sec), abrupt onset/offset, no post-ictal confusion; usually children; can occur 100+/day | 3 Hz generalized spike-and-wave (hallmark) |
| Myoclonic | Brief shock-like jerks, often in the morning; consciousness preserved | Generalized polyspike-and-wave |
| Atonic ("drop attacks") | Sudden loss of muscle tone → fall; very brief; high injury risk | Generalized spike-and-wave or polyspike |
| Tonic | Sustained stiffening without clonic phase; common in Lennox-Gastaut syndrome | Generalized fast activity or electrodecremental |
| Clonic | Rhythmic jerking without preceding tonic phase | Generalized spike-and-wave |
10 Status Epilepticus Epilepsy
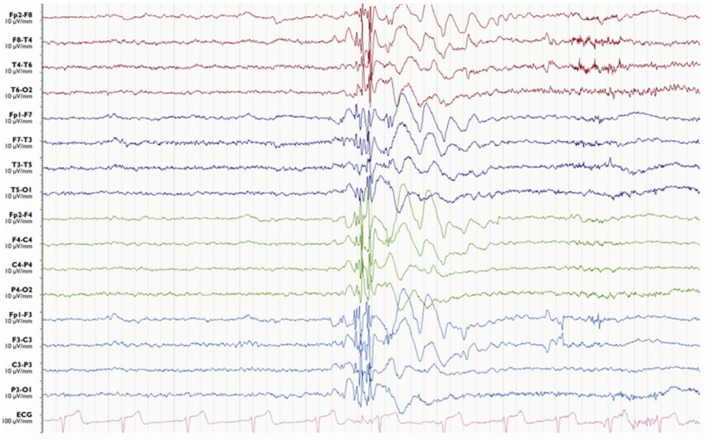
Defined as a seizure lasting ≥ 5 minutes or two or more seizures without return to baseline between them. Convulsive status epilepticus (CSE) is a life-threatening emergency — mortality is 10–20%; risk of permanent neuronal injury increases with duration. Refractory status epilepticus: seizures continue despite two adequate AED trials. Super-refractory: persists ≥ 24 hours despite anesthesia.
Stepwise Treatment Protocol
| Phase | Time | Agent | Dose |
|---|---|---|---|
| 1. Stabilization (emergent) | 0–5 min | Benzodiazepine (first-line) | Lorazepam (Ativan) 0.1 mg/kg IV (max 4 mg/dose, may repeat ×1); OR Midazolam 10 mg IM (if no IV access — RAMPART trial); OR Diazepam 0.15 mg/kg IV (max 10 mg) |
| 2. Urgent control | 5–20 min | Second-line AED | Fosphenytoin 20 mg PE/kg IV (max rate 150 mg PE/min); OR Levetiracetam (Keppra) 60 mg/kg IV (max 4500 mg) over 15 min; OR Valproate (Depakote) 40 mg/kg IV (max 3000 mg) over 10 min. The ESETT trial (2019) showed equivalent efficacy among these three |
| 3. Refractory | 20–60 min | Repeat second-line OR third-line | If not tried: use another second-line agent. If still seizing: proceed to continuous infusion |
| 4. Refractory — anesthesia | > 60 min | Continuous IV anesthesia | Midazolam drip (0.2 mg/kg bolus, then 0.05–2 mg/kg/hr); OR Propofol (2 mg/kg bolus, then 1–5 mg/kg/hr — watch for propofol infusion syndrome); OR Pentobarbital (5 mg/kg bolus, then 1–5 mg/kg/hr — for super-refractory). Titrate to EEG burst-suppression |
Continuous or near-continuous seizure activity without overt convulsive movements. Presents as prolonged confusion, altered consciousness, behavioral change, or subtle findings (eye deviation, nystagmoid jerks, facial twitching). Diagnosed only by EEG. Must be considered in any patient with unexplained altered mental status, especially after convulsive SE when the patient "does not wake up." Up to 8% of comatose ICU patients have NCSE on continuous EEG monitoring.
11 Epilepsy Syndromes & AED Selection Epilepsy
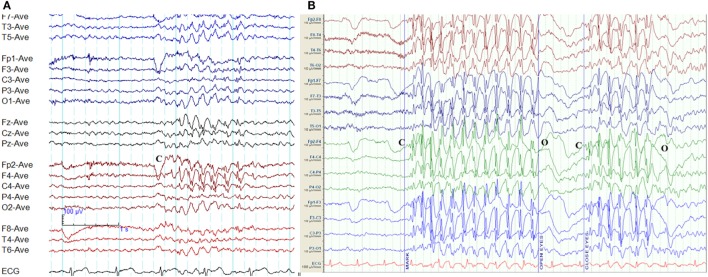
Key Epilepsy Syndromes
| Syndrome | Age of Onset | Seizure Types | EEG | First-Line AED |
|---|---|---|---|---|
| Childhood absence epilepsy | 4–10 years | Absence (multiple daily) | 3 Hz spike-and-wave, hyperventilation provoked | Ethosuximide (absence only) or valproate |
| Juvenile myoclonic epilepsy (JME) | 12–18 years | Myoclonic (morning), generalized tonic-clonic, absence | 4–6 Hz polyspike-and-wave | Valproate (males), levetiracetam or lamotrigine (females — valproate teratogenicity) |
| Temporal lobe epilepsy | Any age | Focal impaired awareness (aura → staring, automatisms) | Temporal sharp waves/spikes | Carbamazepine, oxcarbazepine, lamotrigine, levetiracetam |
| Lennox-Gastaut syndrome | 1–7 years | Tonic, atonic, atypical absence, tonic-clonic | Slow (< 2.5 Hz) spike-and-wave | Valproate, clobazam, rufinamide, cannabidiol (Epidiolex) |
| West syndrome (infantile spasms) | 3–12 months | Epileptic spasms (clusters) | Hypsarrhythmia | ACTH/vigabatrin |
| Benign rolandic epilepsy (self-limited epilepsy with centrotemporal spikes) | 3–13 years | Focal motor (face/arm), often nocturnal | Centrotemporal spikes | Often no treatment needed; remits by adolescence |
AED Selection by Seizure Type
| Seizure Type | First-Line Options | Avoid |
|---|---|---|
| Focal (any type) | Carbamazepine, oxcarbazepine, lamotrigine, levetiracetam, lacosamide | Ethosuximide (ineffective) |
| Generalized tonic-clonic | Valproate, lamotrigine, levetiracetam | Carbamazepine, phenytoin (may worsen) |
| Absence | Ethosuximide, valproate | Carbamazepine, phenytoin, gabapentin (worsen) |
| Myoclonic | Valproate, levetiracetam | Carbamazepine, phenytoin, gabapentin, lamotrigine (may worsen) |
| Atonic | Valproate, clobazam, lamotrigine | Carbamazepine (may worsen) |
12 Alzheimer's Disease Neurodegenerative
Alzheimer's disease (AD) is the most common cause of dementia, accounting for 60–70% of cases. Pathologically defined by extracellular amyloid-beta (Aβ) plaques and intracellular neurofibrillary tangles (hyperphosphorylated tau protein). These begin accumulating 15–20 years before clinical symptoms. The earliest affected region is the entorhinal cortex/hippocampus, explaining why short-term memory loss is the hallmark presenting symptom.
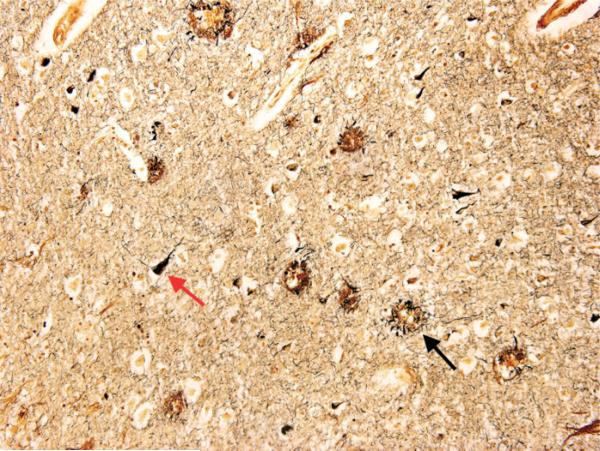
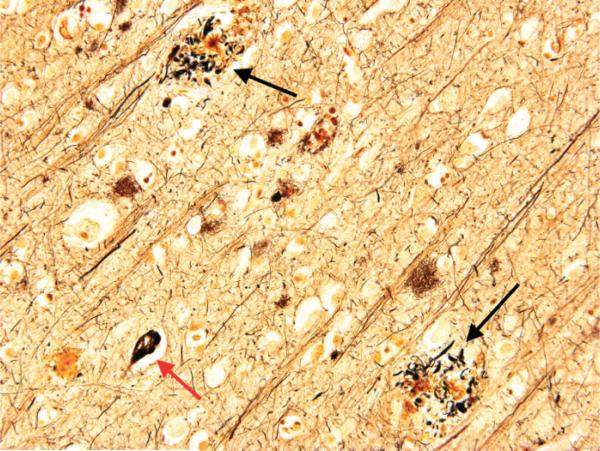
Clinical progression: Mild cognitive impairment (MCI) due to AD → mild AD (instrumental ADLs affected — finances, medications, cooking) → moderate AD (basic ADLs impaired — dressing, bathing; behavioral symptoms emerge — agitation, delusions, wandering) → severe AD (non-verbal, bed-bound, dysphagia, incontinence). Average survival from diagnosis: 8–10 years.
Diagnosis: Clinical criteria (NIA-AA 2011): insidious onset, progressive amnestic (typical) or non-amnestic (atypical — posterior cortical atrophy/logopenic aphasia) cognitive decline, exclusion of other causes. Biomarkers: CSF (low Aβ42, elevated phospho-tau and total tau), amyloid PET (florbetapir, flutemetamol — positive in AD), tau PET (flortaucipir), and MRI showing medial temporal lobe/hippocampal atrophy disproportionate to age.
Treatment: Cholinesterase inhibitors (mild-moderate): donepezil (Aricept) 5–10 mg daily, rivastigmine (Exelon) patch 4.6–13.3 mg/24hr, galantamine (Razadyne) 8–24 mg daily — modest symptomatic benefit, do not modify disease course. Memantine (Namenda) 10 mg BID (NMDA receptor antagonist) for moderate-severe AD — can be combined with a cholinesterase inhibitor. Anti-amyloid monoclonal antibodies: lecanemab (Leqembi) 10 mg/kg IV biweekly — slowed cognitive decline by 27% over 18 months in the Clarity AD trial; aducanumab (Aduhelm) — controversial accelerated FDA approval; donanemab — targets N-terminal truncated Aβ. Major risk: ARIA (amyloid-related imaging abnormalities) — cerebral edema and/or microhemorrhages, highest in APOE4 homozygotes.
13 Parkinson's Disease Neurodegenerative
Parkinson's disease (PD) is the second most common neurodegenerative disorder. Pathology: loss of dopaminergic neurons in the substantia nigra pars compacta with intraneuronal Lewy bodies (alpha-synuclein aggregates). Symptoms appear when ~60–80% of striatal dopamine is depleted. Mean age of onset: 60 years; prevalence increases with age.
Cardinal Motor Features
The diagnosis requires bradykinesia (slowness and decrement in amplitude/speed of repetitive movements — the core required feature) PLUS at least one of: (1) rest tremor — 4–6 Hz, "pill-rolling," asymmetric at onset, diminishes with action; (2) rigidity — lead-pipe or cogwheel (rigidity + superimposed tremor), present throughout passive range of motion. Postural instability appears later and marks a transition to higher fall risk.
Hoehn & Yahr Staging
| Stage | Description |
|---|---|
| 1 | Unilateral involvement only |
| 1.5 | Unilateral + axial involvement |
| 2 | Bilateral involvement without impairment of balance |
| 2.5 | Mild bilateral disease with recovery on pull test |
| 3 | Mild-moderate bilateral disease; some postural instability; physically independent |
| 4 | Severe disability; still able to walk/stand unassisted |
| 5 | Wheelchair-bound or bedridden unless aided |
Non-Motor Features (Often Precede Motor Symptoms by Years)
REM sleep behavior disorder (RBD — acting out dreams), hyposmia (reduced sense of smell), constipation, depression/anxiety, orthostatic hypotension, urinary urgency, cognitive impairment (PD dementia develops in ~80% over the disease course). RBD + hyposmia + constipation together strongly predict future PD or Lewy body dementia.
Pharmacotherapy
| Drug (Brand) | Mechanism | Role | Key Pearl |
|---|---|---|---|
| Carbidopa/Levodopa (Sinemet) | Levodopa = dopamine precursor; carbidopa = peripheral DDC inhibitor (prevents peripheral conversion → reduces nausea) | Most effective PD drug — gold standard | Motor fluctuations develop after 5–10 years ("wearing off," ON/OFF, dyskinesias); take 30 min before meals (protein competes for absorption) |
| Pramipexole (Mirapex), Ropinirole (Requip) | Dopamine agonists (D2/D3) | Early PD monotherapy (younger patients) or adjunct | Impulse control disorders (gambling, hypersexuality, compulsive shopping) in ~15%; excessive daytime sleepiness |
| Rasagiline (Azilect), Selegiline (Eldepryl) | MAO-B inhibitors | Early monotherapy (mild symptoms) or adjunct to reduce "off" time | Avoid meperidine and tramadol (serotonin syndrome risk); tyramine-related hypertensive crisis is rare at selective MAO-B doses |
| Entacapone (Comtan) | COMT inhibitor | Adjunct to levodopa — extends half-life, reduces "off" time | Available as combination tablet with carbidopa/levodopa (Stalevo); may worsen dyskinesias; orange urine |
| Amantadine (Gocovri) | NMDA antagonist + dopamine reuptake inhibitor | Mild early benefit; main role is reducing levodopa-induced dyskinesias | Livedo reticularis, peripheral edema, hallucinations; renally cleared — dose reduce in CKD |
| Trihexyphenidyl, Benztropine | Anticholinergics | Tremor-predominant PD in younger patients | Avoid in elderly (cognitive worsening, urinary retention, constipation); limited use in modern practice |
Deep Brain Stimulation (DBS): Bilateral stimulation of the subthalamic nucleus (STN) or globus pallidus interna (GPi) for patients with motor fluctuations refractory to medication optimization. Reduces "off" time by ~60%, reduces dyskinesias (especially GPi), and allows levodopa dose reduction. Patient selection: good levodopa responsiveness (predicts DBS benefit), no significant cognitive impairment or uncontrolled psychiatric disease, adequate surgical fitness.
14 Lewy Body Dementia & Frontotemporal Dementia Neurodegenerative
Dementia with Lewy Bodies (DLB)
Second most common neurodegenerative dementia after AD. Core features (2 of 4 = probable DLB): (1) fluctuating cognition with pronounced variations in attention/alertness, (2) recurrent visual hallucinations (detailed, well-formed — people, animals), (3) REM sleep behavior disorder, (4) spontaneous parkinsonism. The "1-year rule" distinguishes DLB from Parkinson's disease dementia: if dementia develops ≥ 1 year after motor symptoms → PD dementia; if dementia precedes or occurs within 1 year of motor symptoms → DLB. DaTscan (ioflupane SPECT) shows reduced dopamine transporter uptake in the striatum, supporting the diagnosis. Critical management consideration: DLB patients are exquisitely sensitive to neuroleptics — typical antipsychotics (haloperidol) can cause severe, potentially fatal parkinsonism and neuroleptic malignant syndrome. If antipsychotics are needed, use low-dose quetiapine or pimavanserin (Nuplazid — selective 5-HT2A inverse agonist, FDA-approved for PD psychosis).
Frontotemporal Dementia (FTD)
A group of neurodegenerative disorders with predominant frontal and anterior temporal atrophy. Typically younger onset (45–65 years) than AD. Three main clinical variants:
| Variant | Core Features | Pathology |
|---|---|---|
| Behavioral variant FTD (bvFTD) | Early personality change: disinhibition, apathy, loss of empathy, perseverative/compulsive behaviors, hyperorality, executive dysfunction with relative preservation of memory and visuospatial function | ~45% tau (Pick bodies), ~50% TDP-43 |
| Semantic variant PPA (svPPA) | Progressive loss of word meaning and object knowledge; fluent but empty speech, surface dyslexia, preserved repetition | Predominantly TDP-43 |
| Non-fluent/agrammatic PPA (nfvPPA) | Effortful, halting speech with grammatical errors; speech apraxia; preserved single-word comprehension | Predominantly tau (4R tauopathy) |
There is no disease-modifying treatment. Management is symptomatic: SSRIs for compulsive behaviors and irritability; trazodone for agitation; no benefit from cholinesterase inhibitors (unlike AD). FTD-ALS overlap: ~15% of FTD patients develop ALS features (C9orf72 hexanucleotide repeat expansion is the most common shared genetic cause).
15 Amyotrophic Lateral Sclerosis (ALS) Neurodegenerative
ALS is the most common motor neuron disease — progressive degeneration of both upper motor neurons (cortex → corticospinal tract) and lower motor neurons (anterior horn cells → peripheral nerves). Incidence: ~2/100,000 per year. Median survival from symptom onset: 3–5 years. Death is usually from respiratory failure. ~10% of cases are familial (SOD1, C9orf72, FUS, TARDBP mutations).
Presentation: Painless, asymmetric limb weakness is the most common onset (~70%) — often distal hand wasting with fasciculations. Bulbar onset (~25%) presents with dysarthria, dysphagia, and tongue fasciculations/atrophy. The hallmark is concurrent UMN signs (hyperreflexia, spasticity, Babinski) and LMN signs (atrophy, fasciculations, weakness) in the same body region — a combination virtually pathognomonic for ALS. Sensation is spared. Eye movements and sphincter function are typically preserved until late stages.
Diagnosis: Primarily clinical, supported by EMG/NCS (widespread active denervation with reinnervation in ≥ 3 body regions; normal sensory conduction). The revised El Escorial criteria require UMN + LMN signs in multiple regions with progressive spread and exclusion of mimics (cervical myelopathy, multifocal motor neuropathy, inclusion body myositis). MRI of brain and spine to exclude structural causes.
Management: Riluzole (Rilutek) 50 mg BID — glutamate antagonist, extends survival by ~2–3 months; the only drug with proven survival benefit until recently. Edaravone (Radicava) — free radical scavenger; slowed functional decline in a subgroup of early ALS patients. Tofersen (Qalsody) — antisense oligonucleotide for SOD1-ALS specifically. Multidisciplinary care: pulmonology (noninvasive ventilation — BiPAP extends survival by months and improves quality of life), nutrition (PEG placement before FVC drops below 50%), speech therapy, PT/OT, palliative care. Discussions about advance directives and tracheostomy/ventilator decisions should occur early.
16 Huntington's Disease Neurodegenerative
Autosomal dominant trinucleotide repeat disorder — CAG expansion in the huntingtin gene (HTT) on chromosome 4. Normal: < 27 CAG repeats; intermediate: 27–35 (premutation); reduced penetrance: 36–39; full penetrance: ≥ 40. The age of onset inversely correlates with repeat length (genetic anticipation — longer repeats in successive generations, especially with paternal inheritance). Usual onset: 30–50 years.
Clinical triad: (1) Chorea — involuntary, irregular, non-repetitive, flowing movements; the most recognized feature; progresses to dystonia and rigidity in later stages. (2) Behavioral/psychiatric symptoms — depression, irritability, apathy, obsessive-compulsive features; often precede motor symptoms by years. (3) Cognitive decline — subcortical pattern (executive dysfunction, psychomotor slowing, impaired attention) progressing to dementia. Juvenile-onset (Westphal variant, ≥ 60 repeats): predominantly rigid-akinetic rather than choreic.
Diagnosis: Genetic testing (CAG repeat count) is definitive. Imaging: caudate nucleus atrophy with ventricular enlargement ("boxcar ventricles") on MRI. Pre-symptomatic genetic testing involves extensive genetic counseling due to psychological and insurance implications. Treatment: Chorea: tetrabenazine (Xenazine) or deutetrabenazine (Austedo) — VMAT2 inhibitors that deplete presynaptic dopamine; can cause depression and suicidality (black box warning). No disease-modifying therapy is available. Median survival: 15–20 years from diagnosis.
17 Multiple Sclerosis Demyelinating
MS is the most common demyelinating disease of the CNS, affecting ~1 million people in the US. It is an autoimmune-mediated inflammatory demyelination of the brain and spinal cord white matter, causing axonal damage and progressive neurological disability. Peak onset: 20–40 years; female:male ratio ~3:1. Prevalence increases with latitude (vitamin D hypothesis).
Clinical Course Subtypes
| Subtype | Description | Frequency |
|---|---|---|
| Relapsing-remitting (RRMS) | Discrete attacks (relapses) with full or partial recovery; stable between relapses | ~85% at onset |
| Secondary progressive (SPMS) | Initially RRMS, then gradual progression with or without superimposed relapses | ~50% of RRMS convert within 15–20 years |
| Primary progressive (PPMS) | Gradual progression from onset without discrete relapses | ~15% at onset |
| Progressive-relapsing (PRMS) | Progressive from onset with superimposed acute relapses | Rare (~5%); now considered a variant of PPMS |
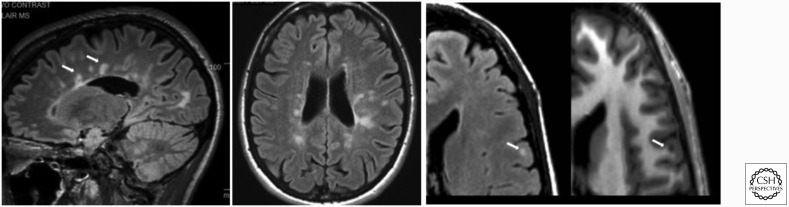
Common Presentations
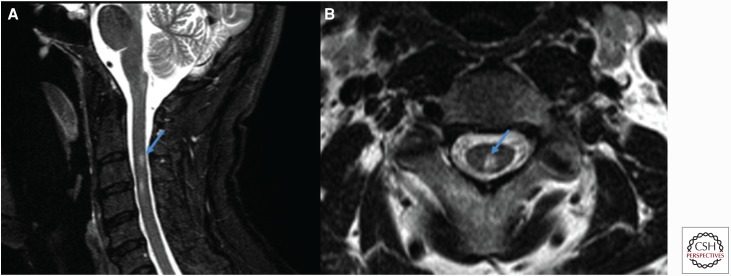
Optic neuritis — unilateral painful vision loss (pain with eye movement), central scotoma, APD; most common presenting symptom (~25%). Transverse myelitis — partial (asymmetric) cord syndrome; sensory level, weakness, bowel/bladder dysfunction; MS-related lesions are typically < 2 vertebral segments (vs NMOSD ≥ 3). Internuclear ophthalmoplegia (INO) — bilateral INO in a young patient is highly suggestive of MS. Lhermitte's sign — electric shock sensation down the spine with neck flexion (cervical cord demyelination). Uhthoff's phenomenon — worsening of symptoms with heat/exercise (temperature-dependent conduction block in demyelinated fibers — not a true relapse).
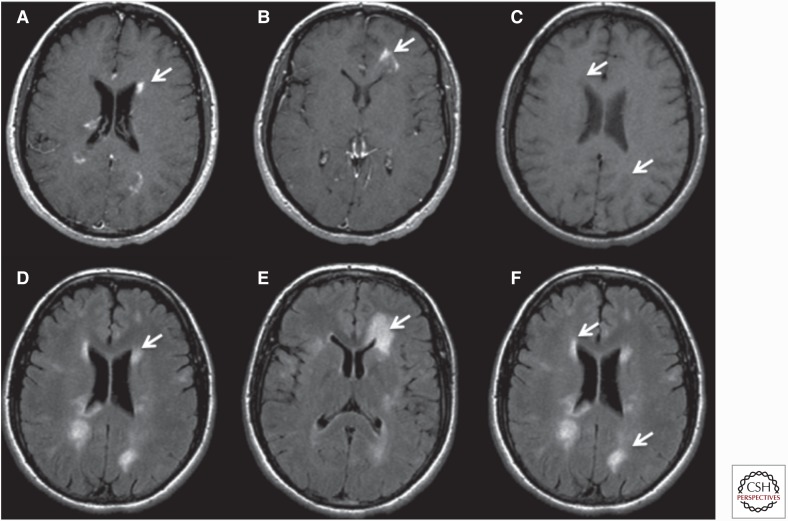
McDonald Criteria (2017 Revision) — Dissemination in Space and Time
MS diagnosis requires demonstration of CNS demyelination disseminated in space (DIS) — lesions in ≥ 2 of 4 typical locations (periventricular, cortical/juxtacortical, infratentorial, spinal cord) — AND disseminated in time (DIT) — simultaneous presence of gadolinium-enhancing and non-enhancing lesions on a single MRI, or a new T2/enhancing lesion on follow-up MRI, or CSF-specific oligoclonal bands (2017 revision allows OCBs to substitute for DIT). This means a single MRI plus positive CSF OCBs can now establish the diagnosis without waiting for a second clinical attack.
Disease-Modifying Therapies (DMTs)
| Drug (Brand) | Route | Mechanism | Efficacy Tier | Key Risk |
|---|---|---|---|---|
| Interferon beta-1a (Avonex, Rebif) | IM / SC | Immunomodulation | Moderate | Flu-like symptoms, injection site reactions, LFT elevation |
| Glatiramer acetate (Copaxone) | SC | Myelin basic protein analog; shifts T-cell response | Moderate | Injection site reactions, lipoatrophy |
| Dimethyl fumarate (Tecfidera) | PO | Nrf2 activation; anti-inflammatory/antioxidant | Moderate | GI symptoms, flushing, lymphopenia, rare PML |
| Teriflunomide (Aubagio) | PO | Dihydroorotate dehydrogenase inhibitor (pyrimidine synthesis) | Moderate | Hepatotoxicity, teratogenic (requires cholestyramine washout), hair thinning |
| Fingolimod (Gilenya) | PO | S1P receptor modulator; traps lymphocytes in lymph nodes | Moderate-High | First-dose bradycardia (6-hour monitoring), macular edema, PML, VZV reactivation |
| Natalizumab (Tysabri) | IV q4w | Anti-alpha-4-integrin; blocks lymphocyte CNS migration | High | PML (especially JCV antibody-positive; index > 1.5 = higher risk); monthly infusion |
| Ocrelizumab (Ocrevus) | IV q6mo | Anti-CD20; B-cell depletion | High | Infusion reactions, increased infection risk, potential malignancy signal; first DMT approved for PPMS |
| Alemtuzumab (Lemtrada) | IV (annual courses) | Anti-CD52; lymphocyte depletion | High | Secondary autoimmunity (thyroid 30–40%, ITP, anti-GBM disease); REMS program |
| Cladribine (Mavenclad) | PO (annual courses) | Purine nucleoside analog; lymphocyte depletion | High | Lymphopenia, herpes zoster reactivation, teratogenic |
Acute relapse management: IV methylprednisolone 1 g/day for 3–5 days (speeds recovery from relapses but does not change long-term disability). Plasma exchange (PLEX) for steroid-refractory relapses (5–7 exchanges over 10–14 days).
18 Neuromyelitis Optica Spectrum Disorder (NMOSD) Demyelinating
NMOSD is an autoimmune astrocytopathy (not a variant of MS). ~75% of patients are seropositive for aquaporin-4 IgG (AQP4-IgG) antibodies, which target AQP4 water channels on astrocyte foot processes concentrated at the optic nerve, spinal cord, and area postrema. Female predominance (~9:1). More common in Black, Asian, and Hispanic populations compared to MS.
Core clinical syndromes: (1) Optic neuritis — often severe, bilateral, with poor recovery (unlike MS). (2) Longitudinally extensive transverse myelitis (LETM) — ≥ 3 vertebral segments (unlike MS: typically < 2). (3) Area postrema syndrome — intractable nausea, vomiting, hiccups (from dorsal medullary involvement).
Distinguishing NMOSD from MS: NMOSD has AQP4-IgG positivity, long cord lesions, severe optic neuritis, brain MRI often initially normal or with atypical lesions (area postrema, periependymal), CSF typically shows neutrophilic pleocytosis (not lymphocytic) without oligoclonal bands. Treatment: Acute attacks: high-dose IV steroids + PLEX (PLEX is more commonly needed than in MS). Prevention: eculizumab (Soliris — complement C5 inhibitor), inebilizumab (Uplizna — anti-CD19), satralizumab (Enspryng — anti-IL-6R) — all FDA-approved for AQP4+ NMOSD. Rituximab (off-label, anti-CD20) is also widely used. Azathioprine and mycophenolate are alternative options. MS DMTs (interferon-beta, fingolimod, natalizumab) can worsen NMOSD — accurate diagnosis is critical.
19 Guillain-Barré Syndrome Autoimmune
GBS is an acute, immune-mediated polyradiculoneuropathy — the most common cause of acute flaccid paralysis. Incidence: ~1–2/100,000/year. Typically follows an infection by 1–4 weeks (Campylobacter jejuni gastroenteritis is the most common trigger — ~30%; also CMV, EBV, Mycoplasma, Zika, influenza, and post-vaccination). Pathogenesis: molecular mimicry — antibodies against microbial antigens cross-react with gangliosides on peripheral nerve myelin or axons.
Clinical Subtypes
| Subtype | Target | Features | Antibody |
|---|---|---|---|
| Acute inflammatory demyelinating polyradiculoneuropathy (AIDP) | Myelin | Most common in Western countries (~85%); ascending weakness, areflexia, sensory symptoms, albuminocytologic dissociation in CSF | Not routinely tested |
| Acute motor axonal neuropathy (AMAN) | Motor axons | More common in Asia; pure motor, often post-Campylobacter; can be severe | Anti-GM1, anti-GD1a |
| Miller Fisher syndrome | CN III, IV, VI + peripheral nerves | Triad: ophthalmoplegia + ataxia + areflexia; descending (unlike typical GBS) | Anti-GQ1b (~90%) |
Presentation: Symmetric ascending weakness (legs before arms) over days to 2–4 weeks, areflexia, back/limb pain. Nadir within 4 weeks. Respiratory failure occurs in ~25% (monitor FVC q4–6h: intubate if FVC < 20 mL/kg or decreasing trend, NIF < -30 cmH2O — the "20/30 rule"). Autonomic dysfunction (blood pressure lability, tachycardia, arrhythmias, urinary retention) can be life-threatening.
Diagnosis: CSF: albuminocytologic dissociation (elevated protein with normal WBC) — may be normal in the first week. NCS/EMG: demyelinating pattern (prolonged distal latencies, conduction block, slowed velocities, prolonged F-waves) in AIDP; axonal pattern (reduced CMAP amplitudes with normal velocities) in AMAN. Treatment: IVIG (0.4 g/kg/day × 5 days) or plasma exchange (5 exchanges over 1–2 weeks) — equally effective; combining them adds no benefit. Corticosteroids are NOT effective in GBS (unlike most autoimmune neurological conditions).
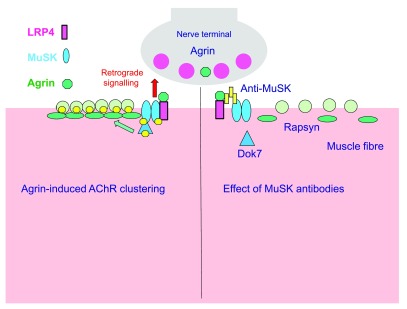
20 Myasthenia Gravis Autoimmune
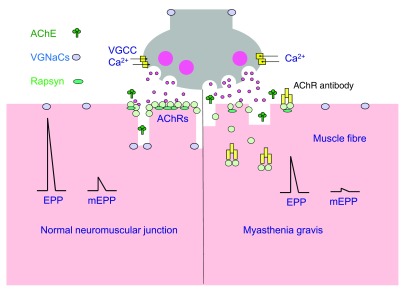
MG is an autoimmune disorder of the neuromuscular junction. ~85% have antibodies against the acetylcholine receptor (AChR) on the postsynaptic membrane; ~5–8% have anti-MuSK (muscle-specific kinase) antibodies; ~5–10% are "seronegative" (may have anti-LRP4 or low-titer AChR). Bimodal age distribution: women 20–30, men 60–70. Strong association with thymic abnormalities: 65% thymic hyperplasia, 10–15% thymoma.
Presentation: Fluctuating, fatigable weakness — worse with use, better with rest. Ocular MG (~50% at onset): ptosis (often asymmetric) and diplopia. Generalized MG: spreads to bulbar (dysphagia, dysarthria, facial weakness), limb (proximal > distal), and respiratory muscles. Myasthenic crisis: respiratory failure from weakness (FVC < 15–20 mL/kg); triggers include infection, surgery, medication changes, or initiation of corticosteroids ("steroid dip"). Anti-MuSK MG has a distinct phenotype: more prominent facial, bulbar, and respiratory weakness; less ocular involvement; poor response to cholinesterase inhibitors.
Diagnosis: AChR antibodies (positive in 85% of generalized, 50% of pure ocular). Ice test (place ice pack on ptotic eyelid for 2 minutes — improvement suggests MG; cooling improves NMJ function). Edrophonium (Tensilon) test — rarely used now due to cardiac risks. Repetitive nerve stimulation: decrement of ≥ 10% in CMAP amplitude at 3 Hz (low-frequency stimulation). Single-fiber EMG: increased jitter — most sensitive test for MG (~95%).
Treatment: Pyridostigmine (Mestinon) 60 mg PO TID (acetylcholinesterase inhibitor — symptomatic). Immunosuppression: prednisone (start low, titrate up — watch for initial worsening), steroid-sparing agents (azathioprine, mycophenolate, tacrolimus). Thymectomy — for all thymoma patients; also beneficial in AChR+ generalized MG even without thymoma (MGTX trial). Crisis management: IVIG or PLEX for rapid improvement. Efgartigimod (Vyvgart) — FcRn inhibitor that reduces IgG levels; FDA-approved for generalized MG. Avoid in MG: aminoglycosides, fluoroquinolones, macrolides, magnesium, beta-blockers, neuromuscular blocking agents, D-penicillamine, IV contrast (relative), botulinum toxin.
21 Autoimmune Encephalitis Autoimmune
A group of inflammatory brain diseases caused by antibodies against neuronal surface or intracellular antigens. The most common and treatable form is anti-NMDA receptor encephalitis — the most frequent cause of autoimmune encephalitis, predominantly affecting young women. Associated with ovarian teratoma in ~50% of female patients > 18 years.
Anti-NMDA receptor encephalitis progression: (1) Prodromal viral-like illness → (2) Psychiatric symptoms (psychosis, agitation, hallucinations — often initially misdiagnosed) → (3) Seizures → (4) Movement disorders (orofacial dyskinesias, choreoathetosis) → (5) Autonomic instability (dysautonomia, hypoventilation) → (6) Decreased consciousness (often requiring ICU care). Other antibody-associated syndromes: Anti-LGI1: faciobrachial dystonic seizures, hyponatremia (SIADH); Anti-CASPR2: limbic encephalitis, neuromyotonia, Morvan syndrome; Anti-AMPAR: limbic encephalitis, high tumor association; Anti-GABA-B: limbic encephalitis, seizures, SCLC association.
Diagnosis: CSF antibody testing (send both CSF and serum); CSF may show lymphocytic pleocytosis and/or oligoclonal bands. MRI may be normal or show medial temporal FLAIR signal. EEG often shows diffuse slowing or extreme delta brush pattern (specific for anti-NMDA receptor encephalitis). Treatment: First-line: IV methylprednisolone + IVIG or PLEX. Second-line (if poor response after 2 weeks): rituximab + cyclophosphamide. Tumor removal (teratoma resection) is critical — recovery depends on it. Relapse rate ~12–25% (higher without tumor removal or without adequate immunotherapy).
22 Essential Tremor Movement
Essential tremor (ET) is the most common movement disorder — 8–10 times more common than Parkinson's disease. Bilateral, largely symmetric action tremor (postural and kinetic) at 4–12 Hz, predominantly affecting the hands and arms; may also involve the head ("yes-yes" or "no-no"), voice, and legs. Typically worsens with age. A family history is present in ~50% (autosomal dominant with variable penetrance). Alcohol transiently improves tremor in ~50–70% of patients — a useful historical clue.
ET vs PD tremor: ET is primarily an action/postural tremor (holding arms outstretched, pouring water); PD tremor is primarily at rest (pill-rolling). ET is usually bilateral and symmetric at onset; PD is asymmetric. ET lacks the bradykinesia and rigidity seen in PD. ET may involve head tremor; PD head tremor is rare without limb involvement. However, ~5% of ET patients later develop PD.
Treatment: First-line: propranolol (60–320 mg/day — long-acting preferred) or primidone (25–750 mg at bedtime — start low, metabolized to phenobarbital). Second-line: topiramate, gabapentin, alprazolam. Botulinum toxin injections for head/voice tremor refractory to oral medications. DBS of the ventral intermediate nucleus (VIM) of the thalamus or MRI-guided focused ultrasound thalamotomy for severe, medication-refractory ET.
23 Dystonia Movement
Dystonia is characterized by sustained or intermittent muscle contractions causing abnormal postures and/or repetitive movements. Classified by: (1) body distribution: focal (one region — cervical dystonia, blepharospasm, writer's cramp), segmental (two or more contiguous regions), multifocal (two or more non-contiguous), generalized, hemidystonia (one side — suggests contralateral basal ganglia lesion); (2) age of onset: early (< 26, often generalized, DYT1 gene) vs adult (usually focal). Cervical dystonia (torticollis) is the most common focal dystonia.
Treatment: Focal dystonias: botulinum toxin (onabotulinumtoxinA = Botox, abobotulinumtoxinA = Dysport, incobotulinumtoxinA = Xeomin) injected into affected muscles — first-line and most effective treatment. Generalized dystonia: anticholinergics (trihexyphenidyl — tolerated better in children than adults), baclofen, benzodiazepines. DBS of the GPi for medication-refractory generalized dystonia (especially DYT1-positive — ~60% improvement). Dopa-responsive dystonia (DRD / Segawa disease) — presents in childhood with diurnal fluctuation (worse in evening); dramatic and sustained response to low-dose levodopa; caused by GTP cyclohydrolase 1 deficiency. Always try levodopa in childhood-onset dystonia.
24 Chorea, Tics & Tourette's Movement
Chorea
Involuntary, irregular, purposeless, non-rhythmic movements that flow from one body part to another. Key causes: Huntington's disease (see Section 16), Sydenham's chorea (post-streptococcal, associated with rheumatic fever — "St. Vitus' dance"), chorea gravidarum (pregnancy — often in patients with prior Sydenham's or antiphospholipid syndrome), drug-induced (levodopa, oral contraceptives, phenytoin, stimulants), vascular (subthalamic or caudate stroke), autoimmune (anti-basal ganglia antibodies, SLE). When chorea affects one side: hemiballismus — large-amplitude, flinging, proximal limb movements, classically from a contralateral subthalamic nucleus lesion (often small lacunar infarct in a diabetic patient).
Tics & Tourette Syndrome
Tics are sudden, rapid, recurrent, non-rhythmic motor movements or vocalizations. They are semi-voluntary — preceded by a premonitory urge and temporarily suppressible (unlike chorea). Tourette syndrome requires: onset before age 18; multiple motor tics + ≥ 1 vocal tic (not necessarily concurrent); tics present for > 1 year. Peak tic severity: ages 10–12; ~2/3 of patients improve significantly by adulthood. Common comorbidities: ADHD (~50%), OCD (~30%), anxiety, learning disabilities. Coprolalia (involuntary obscene utterances) occurs in < 15% of Tourette's patients — it is not required for diagnosis and is far less common than popularly believed.
Treatment: Education and watchful waiting if tics are mild. Behavioral therapy: Comprehensive Behavioral Intervention for Tics (CBIT) — first-line non-pharmacological. Medications (for moderate-severe functional impairment): alpha-2 agonists (clonidine, guanfacine — first-line pharmacological; also help ADHD comorbidity), VMAT2 inhibitors (tetrabenazine, deutetrabenazine), antipsychotics (fluphenazine, aripiprazole — effective but risk of tardive dyskinesia with prolonged use). DBS of the GPi for severe, refractory cases.
25 Drug-Induced Movement Disorders Movement
| Disorder | Mechanism | Culprit Drugs | Features | Treatment |
|---|---|---|---|---|
| Acute dystonic reaction | Dopamine receptor blockade | Haloperidol, metoclopramide, prochlorperazine | Hours to days after starting drug; torticollis, oculogyric crisis, trismus | Diphenhydramine 50 mg IV/IM or benztropine 1–2 mg IV/IM — rapid response |
| Akathisia | Dopamine blockade (unclear exact mechanism) | Antipsychotics, metoclopramide, SSRIs | Subjective restlessness, inability to sit still, pacing; can be misdiagnosed as agitation → more antipsychotic → worsening | Reduce/stop offending drug; propranolol, benzodiazepines, mirtazapine |
| Drug-induced parkinsonism | Dopamine receptor blockade | Antipsychotics (all; quetiapine/clozapine lowest risk), metoclopramide, valproate | Symmetric bradykinesia, rigidity, rest tremor; onset within weeks-months | Reduce/switch offending drug; anticholinergics if cannot stop |
| Tardive dyskinesia (TD) | Dopamine receptor upregulation with chronic blockade | Chronic antipsychotic use (≥ 3 months); metoclopramide | Repetitive, stereotyped movements: orofacial (lip smacking, tongue protrusion, chewing) > limbs > trunk; irreversible in ~50% | Stop offending drug if possible; VMAT2 inhibitors: valbenazine (Ingrezza) or deutetrabenazine (Austedo) — FDA-approved for TD |
| Neuroleptic malignant syndrome (NMS) | Severe dopamine blockade | Antipsychotics (especially high-potency), abrupt withdrawal of dopaminergic agents | Hyperthermia, severe rigidity ("lead-pipe"), AMS, autonomic instability, elevated CK (> 1000), leukocytosis; mortality ~10% | Stop offending drug; supportive care (cooling, IVF); dantrolene (muscle relaxant), bromocriptine (dopamine agonist); ICU admission |
| Serotonin syndrome | Excess serotonergic activity | SSRI/SNRI + MAO-I, tramadol, linezolid, triptans | Triad: AMS, autonomic instability, neuromuscular hyperactivity (clonus — especially lower extremity, hyperreflexia, myoclonus); onset within 24h | Stop serotonergic agents; cyproheptadine 12 mg PO load then 2 mg q2h; benzodiazepines; cooling; supportive |
26 Migraine Headache
Migraine is the second most common primary headache (after tension-type) but the most disabling — affects ~15% of the global population; female:male ratio ~3:1. Migraine is a neurovascular disorder involving cortical spreading depression, trigeminal activation, and neurogenic inflammation of meningeal vessels — not merely a "vascular headache."
Diagnostic Criteria (ICHD-3)
Migraine without aura: ≥ 5 attacks lasting 4–72 hours (untreated) with at least 2 of: unilateral, pulsating, moderate-severe intensity, aggravation by routine physical activity; PLUS at least 1 of: nausea/vomiting, photophobia + phonophobia. Migraine with aura: ≥ 2 attacks with fully reversible visual (most common — scintillating scotoma, zigzag lines), sensory (tingling), or language symptoms; aura develops over ≥ 5 minutes, each symptom lasts 5–60 minutes, followed by headache within 60 minutes.
Acute Treatment
| Agent | Dose | Notes |
|---|---|---|
| NSAIDs (ibuprofen, naproxen) | Ibuprofen 400–800 mg; Naproxen 500 mg | First-line for mild-moderate; most effective when taken early |
| Triptans (sumatriptan, rizatriptan, eletriptan) | Sumatriptan 50–100 mg PO, 6 mg SC; Rizatriptan 10 mg | First-line for moderate-severe; 5-HT1B/1D agonists; contraindicated in CAD, uncontrolled HTN, hemiplegic migraine, basilar migraine; avoid within 24h of ergots |
| Gepants (ubrogepant, rimegepant) | Ubrogepant 50–100 mg PO; Rimegepant 75 mg PO/ODT | CGRP receptor antagonists; alternative to triptans; no vasoconstrictive effects; can be used in cardiovascular disease |
| Ditans (lasmiditan) | 50–200 mg PO | 5-HT1F agonist; no vasoconstriction; causes dizziness/sedation; Schedule V; cannot drive for 8 hours |
| Antiemetics (metoclopramide, prochlorperazine) | Metoclopramide 10 mg IV; Prochlorperazine 10 mg IV | Effective as migraine abortives even without prominent nausea; dopamine antagonist mechanism |
Preventive Therapy (Indicated When ≥ 4 Migraine Days/Month)
| Class | Drug | Dose | Pearl |
|---|---|---|---|
| Beta-blockers | Propranolol, metoprolol | Propranolol 40–240 mg/day | First-line; avoid in asthma; also treats ET |
| Antidepressants | Amitriptyline, venlafaxine | Amitriptyline 10–75 mg QHS | Amitriptyline best evidence; also helps insomnia/tension-type HA; anticholinergic side effects |
| Antiepileptics | Topiramate, valproate | Topiramate 50–100 mg/day | Topiramate causes cognitive slowing, kidney stones, weight loss; valproate teratogenic |
| CGRP monoclonal antibodies | Erenumab (Aimovig), fremanezumab (Ajovy), galcanezumab (Emgality), eptinezumab (Vyepti) | Monthly SC injection (erenumab 70–140 mg) or quarterly IV (eptinezumab) | Effective, well-tolerated; constipation with erenumab; high cost; no hepatic/renal dose adjustment |
| OnabotulinumtoxinA (Botox) | 155 units IM q12 weeks (31 fixed sites) | — | FDA-approved only for chronic migraine (≥ 15 headache days/month) |
27 Tension-Type & Cluster Headache Headache
Tension-Type Headache (TTH)
The most common primary headache. Bilateral, pressing/tightening ("band-like") quality, mild-to-moderate intensity, not aggravated by routine physical activity. No nausea/vomiting (mild nausea allowed in chronic TTH). May have photophobia OR phonophobia but not both. Episodic (< 15 days/month) vs chronic (≥ 15 days/month for ≥ 3 months). Treatment: acute — OTC analgesics (acetaminophen 1000 mg, ibuprofen 400 mg); preventive (for chronic TTH) — amitriptyline 10–75 mg QHS (best evidence).
Cluster Headache
The most severe of all primary headaches — sometimes called "suicide headache." Strictly unilateral, orbital/supraorbital/temporal, excruciating pain lasting 15–180 minutes per attack, occurring 1–8 times daily in clusters lasting weeks to months, separated by remission periods of months to years (episodic form). Chronic form: no remission > 3 months. Male predominance (~3:1). Always accompanied by ipsilateral cranial autonomic symptoms: lacrimation, conjunctival injection, nasal congestion/rhinorrhea, ptosis, miosis, periorbital edema, forehead sweating. Patients are restless/agitated during attacks (unlike migraine patients who prefer to lie still in a dark room).
Acute treatment: (1) High-flow oxygen 12–15 L/min via non-rebreather mask for 15 minutes — effective in ~70%; (2) Sumatriptan 6 mg SC (fastest onset) or 20 mg intranasal — response within 10–15 minutes. Prevention: Verapamil (first-line, 240–960 mg/day in divided doses — ECG monitoring required for PR prolongation); short-term bridging with prednisone taper (60 mg × 5 days, then taper over 2–3 weeks) or greater occipital nerve block (GON block — lidocaine/bupivacaine ± steroid). Lithium, galcanezumab (FDA-approved for episodic cluster headache), topiramate are alternative preventives.
28 Secondary Headache Red Flags & Medication Overuse Headache Headache
"SNOOP" Red Flags for Secondary Headache
| Letter | Red Flag | Concern |
|---|---|---|
| S | Systemic symptoms (fever, weight loss, cancer history, HIV, immunosuppression) | Meningitis, CNS infection, metastasis, temporal arteritis |
| N | Neurological symptoms/signs (focal deficit, papilledema, altered consciousness) | Mass lesion, hemorrhage, increased ICP |
| O | Onset sudden/thunderclap (peak within seconds to 1 minute) | SAH, CVT, dissection, RCVS, pituitary apoplexy |
| O | Onset after age 50 (new headache) | Giant cell arteritis, mass lesion; ESR/CRP + temporal artery biopsy if GCA suspected |
| P | Pattern change (progressive worsening, positional — worse lying down or standing) | Mass lesion (worse lying down/morning = raised ICP); spontaneous intracranial hypotension (orthostatic headache, worse upright) |
Medication Overuse Headache (MOH)
Headache occurring ≥ 15 days/month developing as a consequence of regular overuse of acute headache medications for > 3 months. Thresholds: simple analgesics ≥ 15 days/month; triptans, ergots, opioids, or combination analgesics ≥ 10 days/month. MOH is the third most common headache disorder globally. Treatment: withdrawal of the overused medication (abrupt for triptans/ergots; may taper opioids/barbiturates), bridging therapy (naproxen 500 mg BID, corticosteroid taper, or nerve block), and initiation/optimization of preventive therapy. Preventive CGRP monoclonal antibodies have shown efficacy even without withdrawal — a paradigm shift.
29 Peripheral Neuropathy Neuromuscular
Peripheral neuropathy is damage to the peripheral nerves causing weakness, sensory loss, and/or autonomic dysfunction. The most common type is distal symmetric polyneuropathy (DSPN), presenting with stocking-glove numbness and tingling, with the feet affected first (longest axons are most vulnerable — "length-dependent"). The most common cause worldwide is diabetic neuropathy, affecting ~50% of diabetics over their lifetime.
Etiologic Classification
| Category | Examples | Key Features |
|---|---|---|
| Metabolic | Diabetes, uremia, hypothyroidism, B12 deficiency, copper deficiency | Diabetes: painful distal symmetric; B12 deficiency: dorsal column involvement (subacute combined degeneration with myelopathy) |
| Toxic | Alcohol, chemotherapy (cisplatin, vincristine, taxanes, bortezomib), lead, arsenic | Cisplatin: pure sensory, dose-dependent; vincristine: motor-predominant; alcohol: mixed sensorimotor with nutritional deficiency |
| Inflammatory/Immune | GBS (Section 19), CIDP, vasculitic neuropathy, paraproteinemic | CIDP: chronic relapsing/progressive demyelinating polyradiculoneuropathy; like a chronic GBS; responds to IVIG, steroids, PLEX |
| Hereditary | Charcot-Marie-Tooth (CMT) | Most common hereditary neuropathy; CMT1A (PMP22 duplication, demyelinating — high arches, hammer toes, distal leg weakness, "stork legs") |
| Infectious | HIV, hepatitis C, leprosy, Lyme, diphtheria | HIV: distal sensory painful neuropathy; leprosy: most common treatable neuropathy worldwide |
| Monoclonal gammopathy | MGUS, amyloidosis, myeloma, POEMS syndrome | POEMS: Polyneuropathy + Organomegaly + Endocrinopathy + Monoclonal protein + Skin changes; look for osteosclerotic myeloma |
Workup: Labs: fasting glucose/A1c, CBC, CMP, B12, TSH, SPEP with immunofixation, ESR/CRP. If those are unrevealing: HIV, RPR, ANA, anti-Ro/La, paraneoplastic panel, heavy metals. EMG/NCS distinguishes axonal (reduced amplitudes, normal velocities) from demyelinating (slow velocities, prolonged distal latencies, conduction block). Skin biopsy (intraepidermal nerve fiber density) for small fiber neuropathy (burning pain, autonomic symptoms, normal NCS — standard NCS only tests large fibers).
Neuropathic pain treatment: First-line: duloxetine (Cymbalta) 60 mg/day, pregabalin (Lyrica) 150–300 mg/day, gabapentin (Neurontin) 300–3600 mg/day. Second-line: TCAs (amitriptyline, nortriptyline), topical capsaicin 8% patch, lidocaine 5% patch. Avoid opioids as first-line; consider tramadol only for short-term rescue.
30 Myopathies Neuromuscular
Myopathies present with proximal, symmetric weakness (difficulty rising from chairs, climbing stairs, overhead reaching) without sensory loss. Key distinguishing features from neuropathy: proximal > distal weakness, no fasciculations, normal or reduced reflexes (not absent unless severe), elevated CK.
Inflammatory Myopathies
| Type | Key Features | Pathology | Treatment |
|---|---|---|---|
| Dermatomyositis (DM) | Skin findings + proximal weakness; heliotrope rash (violet eyelids), Gottron's papules (knuckles), V-sign, shawl sign. Malignancy screening required (~25% have occult cancer) | Perifascicular atrophy, complement-mediated microangiopathy (perimysial) | Prednisone + steroid-sparing (methotrexate, azathioprine, IVIG, mycophenolate) |
| Polymyositis (PM) | Proximal weakness without skin findings; subacute onset; no facial or ocular involvement | Endomysial CD8+ T-cell infiltration | Prednisone + steroid-sparing (similar to DM) |
| Inclusion body myositis (IBM) | Most common inflammatory myopathy in patients > 50; insidious; finger flexor and quadriceps weakness (unique pattern: distal + proximal); dysphagia. CK mildly elevated or normal | Rimmed vacuoles, endomysial inflammation | Poor response to immunosuppression; IVIG may help dysphagia; physical therapy; no effective disease-modifying therapy |
| Anti-synthetase syndrome | Myositis + ILD + arthritis + mechanic's hands + Raynaud's + fever; anti-Jo-1 most common antibody | Perifascicular necrosis | Aggressive immunosuppression (ILD drives prognosis); rituximab for refractory |
Other Myopathies
Statin myopathy: Most common drug-induced myopathy. Spectrum: myalgia (most common) → myopathy (CK elevated) → rhabdomyolysis (rare). CK should be checked if symptomatic. If CK > 10× ULN, discontinue statin. Hypothyroid myopathy: Proximal weakness, myalgia, elevated CK, delayed relaxation of reflexes; resolves with thyroid replacement. Steroid myopathy: Proximal weakness, atrophy, normal CK (unlike inflammatory myopathies); risk increases with fluorinated corticosteroids (dexamethasone > prednisone).
31 Nerve Entrapment Syndromes Neuromuscular
| Nerve | Entrapment Site | Syndrome | Motor Deficit | Sensory Deficit | Diagnostic Test |
|---|---|---|---|---|---|
| Median | Carpal tunnel | Carpal tunnel syndrome | Thenar atrophy (abductor pollicis brevis); weak thumb opposition | Palmar digits 1–3 and radial half of 4th | NCS: prolonged distal sensory/motor latency across wrist; Tinel's, Phalen's |
| Ulnar | Cubital tunnel (elbow) | Cubital tunnel syndrome | Interossei, hypothenar atrophy; weak finger abduction/adduction; "claw hand" (4th/5th digits) | Digit 5 and ulnar half of 4th, dorsal hand | NCS: slowing across elbow segment |
| Ulnar | Guyon's canal (wrist) | Guyon's canal syndrome | Similar to cubital tunnel motor deficit; dorsal hand sensation spared (dorsal sensory branch exits proximal to wrist) | Palmar digit 5 and ulnar half of 4th | NCS across wrist |
| Radial | Spiral groove (humerus) | "Saturday night palsy" | Wrist drop (wrist/finger extensors), weak brachioradialis | Dorsal hand — first web space ("anatomical snuffbox") | NCS; EMG of radial-innervated muscles |
| Common peroneal (fibular) | Fibular head | Peroneal neuropathy | Foot drop (ankle dorsiflexion and eversion weakness); spared inversion (tibial nerve) | Lateral leg and dorsum of foot | NCS across fibular head; distinguish from L5 radiculopathy (L5 includes hip abduction, inversion) |
| Lateral femoral cutaneous | Inguinal ligament | Meralgia paresthetica | None (pure sensory nerve) | Burning/numbness of anterolateral thigh | Clinical diagnosis; NCS can confirm |
32 Meningitis Infection
Inflammation of the meninges. The classic triad — headache, fever, neck stiffness — is present in only ~45% of bacterial meningitis cases; however, almost all have at least two of four signs: headache, fever, nuchal rigidity, altered mental status. Kernig's sign (resistance/pain on knee extension with hip flexed to 90°) and Brudzinski's sign (involuntary hip flexion when the neck is passively flexed) have high specificity but low sensitivity (~5%).
CSF Profiles by Etiology
| Parameter | Bacterial | Viral (Aseptic) | Fungal / TB |
|---|---|---|---|
| Opening pressure | Elevated (> 30 cmH2O) | Normal or mildly elevated | Elevated |
| WBC (cells/μL) | 1,000–10,000+ | 10–500 | 10–500 |
| Cell type | Neutrophil predominant | Lymphocyte predominant* | Lymphocyte predominant |
| Protein (mg/dL) | > 100 (often 200–500+) | 50–100 | > 100 |
| Glucose (mg/dL) | < 40 (or CSF:serum ratio < 0.4) | Normal (> 45) | Low (< 40) |
| Gram stain positive | ~60–80% | Negative | India ink (Crypto), AFB (TB — low sensitivity ~20%) |
*Early viral meningitis (< 24h) may show neutrophilic predominance before shifting to lymphocytes.
Empiric Treatment — Bacterial Meningitis
| Age Group | Common Organisms | Empiric Antibiotics |
|---|---|---|
| < 1 month | Group B Strep, E. coli, Listeria | Ampicillin + cefotaxime (or gentamicin) |
| 1 month – 50 years | S. pneumoniae, N. meningitidis | Ceftriaxone + vancomycin |
| > 50 years or immunocompromised | S. pneumoniae, Listeria, gram-negatives | Ceftriaxone + vancomycin + ampicillin (for Listeria) |
Dexamethasone 0.15 mg/kg IV q6h × 4 days — give BEFORE or WITH first dose of antibiotics. Proven benefit in pneumococcal meningitis (reduces mortality and hearing loss). Do NOT delay antibiotics for LP — if LP will be delayed, draw blood cultures and start empiric antibiotics + dexamethasone immediately, then LP when feasible.
33 Encephalitis & Brain Abscess Infection
Viral Encephalitis
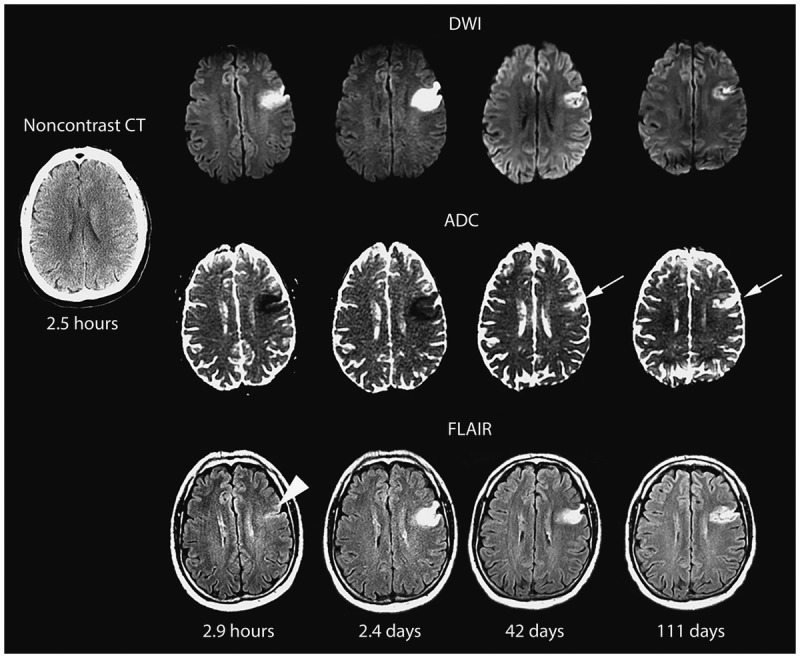
Herpes simplex encephalitis (HSE) is the most common sporadic fatal encephalitis and a neurological emergency. HSV-1 accounts for ~95% of cases (via reactivation in the trigeminal ganglion with retrograde spread to the temporal lobe). Classic presentation: acute fever + altered mental status + focal temporal lobe signs (personality change, olfactory/gustatory hallucinations, aphasia, seizures). MRI shows T2/FLAIR hyperintensity in the medial temporal lobes and insular cortex (often asymmetric). CSF: lymphocytic pleocytosis, elevated protein, normal glucose, HSV PCR positive (sensitivity ~95%). Treatment: acyclovir 10 mg/kg IV q8h for 14–21 days — do NOT wait for PCR results. Untreated mortality is ~70%; with acyclovir, ~20%. Delayed treatment worsens outcomes.
Other viral encephalitides: arboviruses (West Nile, Eastern equine, St. Louis — look for summer/fall, mosquito exposure), CMV (immunocompromised — ventriculitis pattern), VZV, EBV. West Nile: only ~1% of infections cause neuroinvasive disease (encephalitis, meningitis, or acute flaccid paralysis); asymmetric flaccid paralysis mimicking poliomyelitis is characteristic.
Brain Abscess
Encapsulated collection of pus within the brain parenchyma. Sources: contiguous spread (sinusitis → frontal lobe, otitis/mastoiditis → temporal lobe/cerebellum), hematogenous (endocarditis, dental infections, pulmonary AVMs in hereditary hemorrhagic telangiectasia — HHT), post-neurosurgical, penetrating trauma. Most common organisms: mixed aerobic/anaerobic; Streptococci (~70%), Bacteroides, Staphylococci. Immunocompromised: Toxoplasma (HIV + ring-enhancing lesions), Nocardia, Aspergillus.
Presentation: Headache (most common — 70%), focal deficits, fever (only ~50%), seizures. Triad of headache + fever + focal deficit is present in < 50%. Diagnosis: Contrast MRI: ring-enhancing lesion with restricted diffusion on DWI (bright on DWI, dark on ADC — this distinguishes abscess from tumor necrosis/cystic tumor, which typically shows facilitated diffusion). Treatment: Empiric antibiotics: ceftriaxone + metronidazole ± vancomycin (for post-surgical). Aspiration for abscess > 2.5 cm or mass effect. Excision if encapsulated and accessible or no response to aspiration + antibiotics. Duration: 6–8 weeks IV antibiotics.
34 Prion Disease Infection
Prion diseases (transmissible spongiform encephalopathies) are invariably fatal neurodegenerative disorders caused by misfolded prion protein (PrPSc). Creutzfeldt-Jakob disease (CJD) is the most common. Subtypes: sporadic (sCJD — ~85%, mean age 65, no identifiable cause), familial (fCJD — ~10–15%, PRNP gene mutations), iatrogenic (contaminated surgical instruments, dura mater grafts, cadaveric growth hormone), variant CJD (vCJD — acquired from bovine spongiform encephalopathy/"mad cow disease," younger patients, psychiatric onset, later neurological decline).
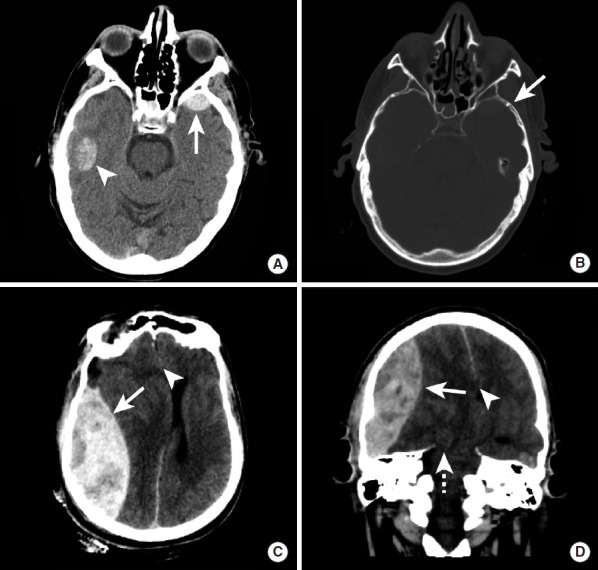
sCJD presentation: Rapidly progressive dementia (weeks to months — NOT months to years like Alzheimer's) with myoclonus ("startle myoclonus"), ataxia, visual symptoms, pyramidal/extrapyramidal signs, akinetic mutism in late stage. Median survival: 5 months from symptom onset. Diagnosis: MRI: cortical ribboning (DWI/FLAIR high signal along the cortical surface) and/or caudate/putamen signal abnormality — highly sensitive (~92%). EEG: periodic sharp wave complexes (PSWCs) at ~1 Hz — present in ~65% but late finding. CSF: RT-QuIC (real-time quaking-induced conversion) assay — sensitivity ~92%, specificity ~99%; elevated 14-3-3 protein (less specific), elevated total tau. Definitive diagnosis requires brain biopsy/autopsy. There is no treatment — care is palliative.
35 Normal Pressure Hydrocephalus Special
NPH is a communicating hydrocephalus (ventricular enlargement without obstruction) presenting with the classic triad: "wet, wacky, and wobbly" — (1) gait disturbance (magnetic/apraxic gait — feet appear glued to the floor, wide-based, short shuffling steps), (2) urinary incontinence (urgency progressing to incontinence), (3) dementia (subcortical pattern — psychomotor slowing, apathy, inattention, executive dysfunction). Gait is typically the earliest and most responsive to treatment. NPH is one of the few reversible causes of dementia.
Diagnosis: MRI: ventriculomegaly out of proportion to sulcal enlargement (Evans' index = frontal horn width / biparietal diameter > 0.3); callosal angle < 90° on coronal sections; periventricular T2 hyperintensity (transependymal CSF flow). Large volume LP (removal of 30–50 mL CSF) — assess gait before and 1–4 hours after; improvement supports diagnosis and predicts shunt response. Continuous lumbar drainage over 72 hours is even more sensitive. Treatment: Ventriculoperitoneal (VP) shunt — improvement in gait (>70%), continence (~50%), cognition (~40%). Programmable shunts allow non-invasive pressure adjustments. Complications: subdural hematoma, infection, shunt malfunction.
36 Idiopathic Intracranial Hypertension Special
Also called pseudotumor cerebri. Elevated intracranial pressure without hydrocephalus or mass lesion. Classic patient: overweight woman of childbearing age. Risk factors: obesity, weight gain, vitamin A excess/retinoids, tetracyclines (doxycycline, minocycline), growth hormone, oral contraceptives (debated).
Presentation: Headache (most common — daily, worse with Valsalva/positional, may be pulsatile), transient visual obscurations (brief graying/blacking out of vision lasting seconds, often with positional change), papilledema (bilateral disc edema on fundoscopy — the hallmark finding), CN VI palsy (false localizing sign from elevated ICP), pulsatile tinnitus ("whooshing" synchronous with heartbeat).
Diagnostic criteria (modified Dandy): (1) Symptoms/signs of raised ICP (papilledema, headache, visual symptoms); (2) LP opening pressure ≥ 25 cmH2O (in lateral decubitus, legs relaxed); (3) Normal CSF composition; (4) No mass, hydrocephalus, or venous sinus thrombosis on imaging (MRI/MRV). Treatment: Weight loss (~5–10% weight loss can resolve the condition). Acetazolamide (Diamox) 500 mg BID, titrate to 1–2 g/day (carbonic anhydrase inhibitor — reduces CSF production; the IIHTT trial confirmed benefit for visual function). Topiramate (secondary — also causes weight loss + reduces CSF production). If vision threatened: optic nerve sheath fenestration or VP/lumboperitoneal shunt. Venous sinus stenting for patients with venous sinus stenosis and pressure gradient.
37 Narcolepsy & Restless Legs Syndrome Special
Narcolepsy
Narcolepsy type 1 (with cataplexy) is caused by autoimmune-mediated destruction of hypocretin/orexin-producing neurons in the lateral hypothalamus, resulting in undetectable CSF hypocretin-1 levels (< 110 pg/mL). Strongly associated with HLA-DQB1*06:02 (present in ~98% of type 1 narcolepsy). Narcolepsy type 2: excessive daytime sleepiness without cataplexy; normal CSF hypocretin.
Pentad of narcolepsy type 1: (1) Excessive daytime sleepiness (the primary and constant symptom — irresistible "sleep attacks"), (2) cataplexy (sudden loss of muscle tone triggered by strong emotions — laughing, surprise — pathognomonic for type 1), (3) sleep paralysis, (4) hypnagogic/hypnopompic hallucinations, (5) disrupted nocturnal sleep. Diagnosis: Overnight polysomnography (to exclude other sleep disorders) followed by multiple sleep latency test (MSLT): mean sleep latency ≤ 8 minutes with ≥ 2 sleep-onset REM periods (SOREMPs). Treatment: Sleepiness: modafinil 100–400 mg/day (first-line), armodafinil, solriamfetol, or traditional stimulants (methylphenidate, dextroamphetamine). Cataplexy: sodium oxybate (Xyrem/Xywav — also improves all symptoms; taken at bedtime + again 2.5–4 hours later), SNRIs (venlafaxine), TCAs (clomipramine), pitolisant (H3 receptor antagonist/inverse agonist).
Restless Legs Syndrome (RLS) / Willis-Ekbom Disease
An urge to move the legs, usually accompanied by uncomfortable sensations (creeping, crawling, aching, tingling), (1) worse at rest, (2) relieved by movement, (3) worse in the evening/night. Affects ~5–10% of adults. Associated with iron deficiency (check ferritin — treat if < 75 ng/mL even if in "normal" range), CKD/dialysis, pregnancy, neuropathy, and dopaminergic medications.
Treatment: Address iron deficiency (oral iron with vitamin C, or IV iron if oral fails or ferritin < 30). Intermittent RLS: low-dose dopamine agonist PRN or gabapentinoid. Chronic moderate-severe: alpha-2-delta ligands (gabapentin enacarbil 600 mg, pregabalin 150–300 mg — preferred first-line to avoid augmentation). Dopamine agonists (pramipexole, ropinirole, rotigotine patch) — effective but risk of augmentation (paradoxical worsening with earlier onset, increased intensity, spread to other body parts with chronic use — occurs in 40–60% within 10 years). Low-dose opioids (oxycodone, methadone) for refractory cases.
38 Neurosarcoidosis Special
Neurological involvement occurs in ~5–10% of sarcoidosis patients but can be the presenting manifestation in ~50% of those affected. Sarcoidosis is a multisystem granulomatous disease of unknown etiology. Neurosarcoidosis can affect any part of the nervous system.
Most common presentations: (1) Cranial neuropathy (~55%) — CN VII (facial palsy, most common; bilateral facial palsy = think sarcoidosis, Lyme, GBS), CN II (optic neuropathy). (2) Leptomeningeal enhancement on MRI (basal predominant). (3) Hypothalamic-pituitary dysfunction (DI, hyperprolactinemia, panhypopituitarism). (4) Myelopathy (longitudinally extensive, can mimic NMOSD). (5) Peripheral neuropathy (small fiber most common). (6) Myopathy (chronic, nodular).
Diagnosis: Biopsy is the gold standard (non-caseating granulomas). CSF: lymphocytic pleocytosis, elevated protein, low glucose (in ~20%), elevated ACE (low sensitivity ~50%). Serum ACE elevated in ~60% (low specificity). Chest imaging (hilar lymphadenopathy in ~90% of sarcoidosis). FDG-PET may identify biopsy-accessible sites. Soluble IL-2 receptor and lysozyme may support diagnosis. Treatment: Corticosteroids (prednisone 0.5–1 mg/kg/day) — first-line; steroid-sparing agents for chronic disease: methotrexate, mycophenolate, azathioprine. Infliximab (anti-TNF-alpha) for refractory neurosarcoidosis — strong evidence. Cyclophosphamide for severe, refractory cases.
39 Lumbar Puncture & CSF Analysis
Indications: Suspected meningitis/encephalitis, SAH (if CT negative), MS workup, NPH evaluation, IIH diagnosis, carcinomatous meningitis, Guillain-Barré. Contraindications: Increased ICP with mass lesion (risk of herniation — get CT first), coagulopathy (INR > 1.5, platelets < 50K), skin infection at puncture site.
Technique: Patient in lateral decubitus or sitting position. Identify L3-L4 or L4-L5 interspace (the spinal cord ends at L1-L2 in adults). Use aseptic technique; insert spinal needle (20 or 22 gauge) with bevel parallel to the longitudinal dural fibers. Measure opening pressure (normal: 6–25 cmH2O in lateral decubitus with legs relaxed). Collect CSF in 4 tubes: (1) cell count + differential, (2) protein + glucose, (3) Gram stain, culture, (4) special studies (cytology, PCR, OCBs, biomarkers).
Normal CSF Values
| Parameter | Normal Range |
|---|---|
| Opening pressure | 6–25 cmH2O |
| Appearance | Clear, colorless |
| WBC | 0–5 cells/μL (lymphocytes/monocytes) |
| RBC | 0 |
| Protein | 15–45 mg/dL |
| Glucose | 50–80 mg/dL (or CSF:serum ratio ≥ 0.6) |
| Oligoclonal bands | Absent (or ≤ 1 band matching serum) |
Post-LP headache: Occurs in ~10–30% — worse upright, relieved lying flat (low-pressure headache from CSF leak at the puncture site). Risk reduced by using atraumatic (Sprotte/Whitacre) needles. Treatment: bed rest, hydration, caffeine (300 mg PO), and autologous epidural blood patch (10–20 mL of patient's blood injected into epidural space — ~90% effective for persistent symptoms).
40 EEG Interpretation
Electroencephalography records cortical electrical activity via scalp electrodes (standard 10-20 system with 21 electrodes). Normal adult background: alpha rhythm (8–13 Hz) predominant over the posterior regions, attenuates with eye opening. Slower rhythms: theta (4–7 Hz) and delta (< 4 Hz) are normal during drowsiness and sleep but abnormal if persistent during wakefulness.
Key EEG Patterns
| Pattern | Description | Clinical Correlation |
|---|---|---|
| 3 Hz spike-and-wave | Generalized, bisynchronous, provoked by hyperventilation | Childhood absence epilepsy |
| 4–6 Hz polyspike-and-wave | Generalized | Juvenile myoclonic epilepsy |
| Hypsarrhythmia | Chaotic, high-amplitude, multifocal spikes and slow waves | West syndrome (infantile spasms) |
| Slow spike-and-wave (< 2.5 Hz) | Generalized | Lennox-Gastaut syndrome |
| Periodic lateralized epileptiform discharges (PLEDs/LPDs) | Lateralized periodic sharp waves | Acute structural lesion (stroke, HSE, tumor); high seizure risk |
| Generalized periodic discharges (GPDs) | Periodic sharp or triphasic waves, bisynchronous | Metabolic encephalopathy (hepatic, renal), CJD, status epilepticus |
| Extreme delta brush | Rhythmic delta activity with superimposed fast beta | Anti-NMDA receptor encephalitis (relatively specific) |
| Burst-suppression | Alternating bursts of activity with flat/suppressed periods | Severe diffuse cerebral dysfunction, therapeutic coma, hypothermia, anesthetic effect |
| Electrocerebral inactivity | No cerebral activity > 2 μV | Brain death (confirmatory test) |
41 EMG / Nerve Conduction Studies
EMG/NCS is the electrodiagnostic study of the peripheral nervous system — the most important test for distinguishing neuropathy from myopathy and for localizing peripheral nerve lesions.
Nerve Conduction Studies (NCS)
Electrically stimulate a peripheral nerve and record the response. Motor NCS: stimulate nerve, record from muscle — generates compound muscle action potential (CMAP). Measures: distal latency, conduction velocity, CMAP amplitude, F-wave latency. Sensory NCS: stimulate and record from the nerve itself — generates sensory nerve action potential (SNAP). Key patterns:
| Pattern | NCS Findings | Interpretation |
|---|---|---|
| Axonal neuropathy | Reduced CMAP/SNAP amplitudes; normal or mildly reduced velocities | Axon loss (diabetes, toxic, most common pattern) |
| Demyelinating neuropathy | Slowed conduction velocities, prolonged distal latencies, conduction block, temporal dispersion; CMAP amplitudes may be preserved initially | Myelin damage (GBS-AIDP, CIDP, CMT1) |
| Entrapment (focal) | Slowing/conduction block localized to the entrapment site; normal above and below | Carpal tunnel, cubital tunnel, peroneal at fibular head |
Electromyography (Needle EMG)
A concentric needle electrode is inserted into muscles to record electrical activity. Three phases of assessment: (1) Insertional activity — brief burst from needle insertion; increased in denervation, decreased in fibrosis/end-stage. (2) Spontaneous activity at rest — normal muscle is electrically silent; fibrillations + positive sharp waves = active denervation; fasciculations = LMN pathology; myotonic discharges = myotonic dystrophy or myotonia. (3) Motor unit action potentials (MUAPs) during voluntary contraction — neuropathic pattern: large, polyphasic, long-duration MUAPs with reduced recruitment (surviving motor units grow larger via reinnervation); myopathic pattern: small, short-duration, polyphasic MUAPs with early recruitment (many weak motor units firing to compensate).
42 Neuroimaging — CT, MRI, MRA & Sequences
CT Applications
| Modality | Indication | Key Findings |
|---|---|---|
| Non-contrast CT head | Acute stroke (rule out hemorrhage), trauma, hydrocephalus | Hyperdense = acute blood; hypodense = edema/infarct (may not be visible < 6–12 hours); dense MCA sign (thrombus) |
| CT angiography (CTA) | LVO detection, carotid stenosis, aneurysm, dissection | Filling defects, vessel occlusion, aneurysm morphology; first-line for acute stroke vascular imaging |
| CT perfusion (CTP) | Ischemic penumbra assessment (extended window thrombectomy) | Core (CBF < 30%) vs penumbra (Tmax > 6 sec); mismatch ratio guides treatment in 6–24 hour window |
| CT venography (CTV) | Cerebral venous thrombosis | Filling defect in venous sinuses; "empty delta sign" (sagittal sinus) |
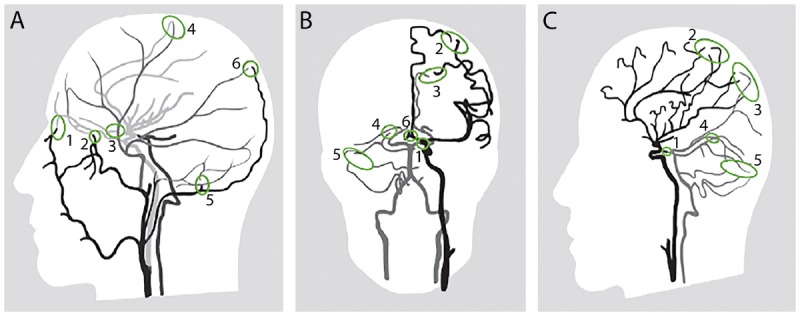
MRI Sequences
| Sequence | What It Shows Best | Key Clinical Uses |
|---|---|---|
| T1-weighted | Anatomy (CSF = dark, fat = bright, gray matter darker than white matter) | Structural anatomy, post-contrast enhancement (gadolinium), fat-containing lesions |
| T2-weighted | Pathology (CSF = bright, edema = bright) | White matter lesions, edema, tumors, inflammation |
| FLAIR (Fluid-Attenuated Inversion Recovery) | Like T2 but CSF signal suppressed → lesions adjacent to ventricles visible | MS plaques (periventricular), cortical infarcts, SAH (leptomeningeal hyperintensity), meningitis |
| DWI (Diffusion-Weighted Imaging) | Restricted diffusion = bright (cytotoxic edema) | Acute ischemic stroke (visible within minutes; high sensitivity/specificity); abscess (restricted diffusion); CJD (cortical ribboning); epidermoid cyst |
| ADC map (Apparent Diffusion Coefficient) | Confirms restricted diffusion (DWI bright + ADC dark = true restriction) | Distinguishes true restriction from "T2 shine-through" (which shows bright on both DWI and ADC) |
| GRE / SWI (Gradient Echo / Susceptibility-Weighted) | Blood products (hemosiderin = dark "blooming") | Microbleeds (CAA, hypertensive), hemorrhagic transformation, cavernous malformations, calcification |
| T1 + Gadolinium contrast | Blood-brain barrier breakdown → enhancement | Active MS lesions, tumors, abscesses (ring-enhancing), leptomeningeal disease, active infection |
| MRA (MR Angiography) | Vascular anatomy (TOF = non-contrast; CE-MRA = with gadolinium) | Intracranial aneurysms, stenosis, dissection, AVM; non-invasive alternative to CTA |
| MR Spectroscopy (MRS) | Metabolite concentrations in tissue | NAA (neuronal marker — decreased in tumors, infarcts); Choline (elevated in tumors — membrane turnover); Lactate (elevated in ischemia, mitochondrial disease) |
43 Classification Systems (All)
| System | Used For | Key Content |
|---|---|---|
| TOAST | Ischemic stroke etiology | 5 categories: large-artery atherosclerosis, cardioembolism, small-vessel, other determined, cryptogenic |
| NIHSS | Ischemic stroke severity | 0–42 scale; 11 items; ≥ 6 suggests LVO; guides tPA/thrombectomy decisions |
| Modified Rankin Scale (mRS) | Stroke functional outcome | 0 = no symptoms; 1 = no significant disability; 2 = slight disability; 3 = moderate (walks without help); 4 = moderately severe (needs help walking); 5 = severe (bedridden); 6 = dead |
| Hunt-Hess | SAH clinical severity | Grades I–V based on clinical presentation (see Section 6) |
| Fisher Scale | SAH CT appearance / vasospasm risk | Grades 1–4 (see Section 6) |
| WFNS Grade | SAH severity (alternative to Hunt-Hess) | Based on GCS and motor deficit; Grade I: GCS 15; II: GCS 13–14 no deficit; III: GCS 13–14 with deficit; IV: GCS 7–12; V: GCS 3–6 |
| ICH Score | ICH 30-day mortality | 0–6 points based on GCS, volume, IVH, infratentorial, age (see Section 6) |
| ABCD2 | Stroke risk after TIA | 0–7 points; Age, BP, Clinical features, Duration, Diabetes |
| CHA2DS2-VASc | Stroke risk in AF | 0–9 points; guides anticoagulation decisions; ≥ 2 (males) or ≥ 3 (females) → anticoagulate |
| HAS-BLED | Bleeding risk on anticoagulation | 0–9 points; HTN, Abnormal renal/liver, Stroke, Bleeding, Labile INR, Elderly, Drugs/alcohol |
| ILAE 2017 | Seizure classification | Focal (aware/impaired awareness) vs Generalized vs Unknown onset; motor vs non-motor |
| Hoehn & Yahr | Parkinson's disease staging | Stages 1–5 based on unilateral/bilateral involvement and functional disability (see Section 13) |
| UPDRS | Parkinson's disease severity | Unified PD Rating Scale; 4 parts: I (mentation/mood), II (ADLs), III (motor exam — most used clinically), IV (complications) |
| EDSS | MS disability | 0.0–10.0 scale (0 = normal; 1–3.5 = ambulatory, minimal disability; 4.0–6.5 = ambulatory with aids; 7.0+ = wheelchair/bedbound; 10.0 = death from MS) |
| McDonald Criteria (2017) | MS diagnosis | Dissemination in space (≥ 2 of 4 typical locations) + dissemination in time (new lesion, simultaneous enhancing/non-enhancing, or CSF OCBs) |
| El Escorial Criteria (revised) | ALS diagnosis | Definite, probable, possible, suspected — based on UMN + LMN signs in body regions |
| Hughes GBS Disability Scale | GBS functional status | 0 = normal; 1 = minor symptoms; 2 = walks 5m unaided; 3 = walks with aid; 4 = chair/bed bound; 5 = ventilated; 6 = dead |
| MGFA Classification | Myasthenia gravis severity | Class I = ocular only; IIA = mild generalized; IIB = moderate generalized; IIIA/B = severe with bulbar; IVA/B = severe with respiratory; V = intubated |
| GCS | Consciousness/coma | Eye (1–4) + Verbal (1–5) + Motor (1–6) = 3–15; ≤ 8 = intubate |
44 Medications Master Table Meds
Antiepileptic Drugs (AEDs)
| Drug (Brand) | Mechanism | Typical Dose | Seizure Types | Key Pearl |
|---|---|---|---|---|
| Levetiracetam (Keppra) | SV2A modulation | 500–1500 mg BID | Broad-spectrum (focal + generalized) | No significant drug interactions; renal dosing; "Keppra rage" behavioral side effect |
| Lamotrigine (Lamictal) | Na+ channel blockade, glutamate inhibition | 100–200 mg BID (slow titration) | Broad-spectrum | SJS risk (especially with valproate co-administration — requires halved dose and slower titration); safe in pregnancy |
| Valproate (Depakote) | Multiple: Na+ channels, GABA enhancement, T-type Ca2+ channels | 500–1500 mg BID | Broad-spectrum (best for generalized) | Category X teratogen (NTDs); hepatotoxic; pancreatitis; weight gain; thrombocytopenia; drug interactions (inhibits metabolism of lamotrigine) |
| Carbamazepine (Tegretol) | Na+ channel blockade | 200–600 mg BID | Focal (can worsen generalized) | Autoinduction (increases own metabolism — recheck levels after 4 weeks); HLA-B*15:02 screening (Asian descent) for SJS; hyponatremia |
| Oxcarbazepine (Trileptal) | Na+ channel blockade | 300–1200 mg BID | Focal | Less autoinduction than carbamazepine; more hyponatremia risk; fewer drug interactions |
| Phenytoin (Dilantin) | Na+ channel blockade | 300–400 mg/day (or fosphenytoin IV) | Focal, tonic-clonic (not absence/myoclonic) | Zero-order kinetics; narrow therapeutic window (total 10–20, free 1–2 mcg/mL); gingival hyperplasia, hirsutism, osteoporosis, cerebellar atrophy |
| Lacosamide (Vimpat) | Slow Na+ channel inactivation | 100–200 mg BID | Focal | Can be loaded IV; well tolerated; PR prolongation (ECG baseline recommended) |
| Topiramate (Topamax) | Multiple: Na+ channels, GABA, glutamate, carbonic anhydrase | 100–200 mg BID | Broad-spectrum | Cognitive dulling ("Dopamax"); weight loss; kidney stones; metabolic acidosis; cleft palate risk in pregnancy |
| Ethosuximide (Zarontin) | T-type Ca2+ channel blockade | 250–750 mg BID | Absence ONLY | First-line for childhood absence epilepsy (if no GTC); GI side effects |
| Brivaracetam (Briviact) | SV2A (higher affinity than levetiracetam) | 50–100 mg BID | Focal (adjunctive) | Less behavioral side effects than levetiracetam; can be loaded IV |
| Clobazam (Onfi) | Benzodiazepine (GABA-A) | 5–20 mg BID | Lennox-Gastaut (adjunctive) | Less sedating than other benzodiazepines; tolerance develops |
| Cannabidiol (Epidiolex) | Uncertain (not CB1/CB2 mediated) | 5–20 mg/kg/day | Dravet, Lennox-Gastaut, tuberous sclerosis | Hepatotoxicity (especially with valproate); somnolence; first FDA-approved cannabis-derived drug |
MS Disease-Modifying Therapies
See Section 17 table for complete MS DMT listing with route, mechanism, efficacy, and key risks.
Parkinson's Disease Drugs
See Section 13 table for complete PD pharmacotherapy with mechanisms, roles, and pearls.
Migraine Medications
See Section 26 tables for acute treatment and preventive therapy.
Stroke Medications
| Drug (Brand) | Mechanism | Dose | Indication | Key Pearl |
|---|---|---|---|---|
| Alteplase (Activase) | Plasminogen activator (tPA) | 0.9 mg/kg IV (max 90 mg) | Acute ischemic stroke within 4.5 hours | 10% bolus, 90% over 60 min; BP < 185/110 before, < 180/105 after |
| Tenecteplase (TNKase) | Modified tPA (longer half-life) | 0.25 mg/kg IV bolus (max 25 mg) | Acute ischemic stroke (LVO, prior to thrombectomy) | Single bolus; increasingly used over alteplase |
| Aspirin | Irreversible COX-1 inhibitor | 81–325 mg daily | Secondary stroke prevention (non-cardioembolic) | Start 24h after tPA; 162–325 mg load in acute stroke if no tPA given |
| Clopidogrel (Plavix) | Irreversible P2Y12 blocker | 75 mg daily | Secondary stroke prevention; DAPT with ASA × 21 days after minor stroke/TIA | CYP2C19 poor metabolizers have reduced benefit; testing may be warranted |
| Apixaban (Eliquis) | Direct factor Xa inhibitor | 5 mg BID (2.5 mg BID if qualifying criteria) | Stroke prevention in non-valvular AF | Preferred DOAC in most AF patients (ARISTOTLE trial); avoid in mechanical valves |
| Nimodipine | Dihydropyridine CCB (cerebrovascular selectivity) | 60 mg PO q4h × 21 days | SAH — prevents delayed cerebral ischemia from vasospasm | ORAL only — IV nimodipine causes severe hypotension; must be PO or via NG tube |
| Nicardipine | Dihydropyridine CCB IV drip | 5 mg/hr, titrate by 2.5 mg/hr q5–15 min (max 15 mg/hr) | BP management in acute stroke / ICH | Smooth, titratable BP control; preferred drip for acute stroke BP management |
Other Neurological Medications
| Drug (Brand) | Mechanism | Indication | Key Pearl |
|---|---|---|---|
| Riluzole (Rilutek) | Glutamate antagonist | ALS | Only ~2–3 month survival benefit; monitor LFTs |
| Pyridostigmine (Mestinon) | Acetylcholinesterase inhibitor | Myasthenia gravis (symptomatic) | Cholinergic crisis if overdosed (SLUDGE: Salivation, Lacrimation, Urination, Defecation, GI distress, Emesis) |
| Donepezil (Aricept) | Acetylcholinesterase inhibitor | Alzheimer's disease (mild-severe) | GI side effects, bradycardia, vivid dreams; benefit is modest and symptomatic |
| Memantine (Namenda) | NMDA receptor antagonist | Alzheimer's disease (moderate-severe) | Can combine with cholinesterase inhibitor; renal dosing required |
| Tetrabenazine (Xenazine) | VMAT2 inhibitor (depletes monoamines) | Huntington's chorea, tardive dyskinesia | Depression/suicidality risk (black box warning); CYP2D6 poor metabolizers need dose reduction |
| Valbenazine (Ingrezza) | VMAT2 inhibitor | Tardive dyskinesia (FDA-approved) | Better tolerated than tetrabenazine; less depression risk; once daily dosing |
| Eculizumab (Soliris) | Anti-C5 complement inhibitor | AQP4+ NMOSD; also generalized MG | Meningococcal vaccination required (≥ 2 weeks before starting); risk of meningococcal infection |
| Sodium oxybate (Xyrem/Xywav) | GABA-B agonist / GHB | Narcolepsy (cataplexy + EDS) | Schedule III controlled substance; REMS program; taken at bedtime + again 2.5–4 hours later; very high sodium content (Xyrem) — Xywav is low-sodium formulation |
45 Abbreviations Master List
Anatomy
Diagnoses
Procedures & Diagnostics
Medications & Labs
Other / General
46 Risk Scores & Screening Tools
GCS (Glasgow Coma Scale): Eye (1–4: none/to pain/to voice/spontaneous) + Verbal (1–5: none/incomprehensible/inappropriate/confused/oriented) + Motor (1–6: none/extension/flexion/withdrawal/localizes/obeys) = 3–15. ≤ 8 = severe TBI, intubation. 9–12 = moderate. 13–15 = mild.
NIHSS: See Section 5. Score 0 = no deficit; 1–4 = minor stroke; 5–15 = moderate; 16–20 = moderate-severe; ≥ 21 = severe.
mRS (Modified Rankin Scale): The primary outcome measure in stroke trials. Ranges from 0 (no symptoms) to 6 (dead). See Section 43 for full scale.
CHA₂DS₂-VASc: CHF (1), HTN (1), Age ≥ 75 (2), DM (1), Stroke/TIA (2), Vascular disease (1), Age 65–74 (1), Sex category female (1) = max 9. Score ≥ 2 in males / ≥ 3 in females → oral anticoagulation for stroke prevention in AF.
HAS-BLED: HTN (1), Abnormal renal/liver function (1 each), Stroke (1), Bleeding history (1), Labile INR (1), Elderly > 65 (1), Drugs/alcohol (1 each) = max 9. Score ≥ 3 = high bleeding risk — does NOT contraindicate anticoagulation but warrants closer monitoring and modifiable risk factor management.
MoCA (Montreal Cognitive Assessment): 30-point screening test. Assesses visuospatial, naming, memory, attention, language, abstraction, orientation. Score ≥ 26 = normal; < 26 = cognitive impairment (more sensitive for MCI than MMSE). Add 1 point if education ≤ 12 years.
EDSS (Expanded Disability Status Scale): MS-specific. 0.0–10.0 in 0.5-point increments. Heavily weighted toward ambulation (scores 4.0–6.5 are almost entirely defined by walking ability). Used in clinical trials and longitudinal MS management.
References & Figure Sources
Figures
- Figures 1–3. Basal ganglia anatomy and motor circuitry. Bhatt D, et al. PMC10435282; Obeso JA, et al. PMC3543080. Open access.
- Figures 4–5. Cerebellum anatomy and functional divisions. Defined S, et al. PMC7055440. Open access.
- Figures 6–7. Spinal cord tracts and Brown-Sequard syndrome. Whelan D, et al. PMC7705369. Open access.
- Figures 8–11, 33, 41–42. Ischemic stroke imaging, thrombectomy, neuroimaging. Defined S, et al. PMC5898964. Open access.
- Figures 12–13, 15, 35–36. Intracranial hemorrhage imaging. An SJ, et al. PMC5307932. Open access.
- Figures 14, 37. Subarachnoid hemorrhage CT imaging. PMC5307932; PMC5619317. Open access.
- Figures 16–18, 40. EEG patterns in epilepsy syndromes. Seneviratne U, et al. PMC5622315. Open access.
- Figures 19–20, 34. Alzheimer disease neuropathology and staging. Perl DP. PMC2918894. Open access.
- Figures 21–23. Parkinson disease pathology and circuits. Obeso JA, et al. PMC3543080; Dickson DW. PMC5718208. Open access.
- Figures 25–27. Multiple sclerosis MRI findings. Filippi M, et al. PMC5932576. Open access.
- Figures 28–29. GBS nerve demyelination and neuromuscular junction in myasthenia gravis. Gilhus NE, et al. PMC4926737. Open access.
- Figures 30–32. Migraine pathophysiology, CSD, and sensitization. Noseda R, Burstein R. PMC3858400. Open access.
- Figure 37–38. NPH and IIH neuroimaging. Defined S, et al. PMC5619317. Open access.
- Figure 39. Spinal needle types for lumbar puncture. Defined S, et al. PMC10295920. Open access.
Key Trials & Guidelines
- Saver JL. Time is brain — quantified. Stroke. 2006;37(1):263-266. PMID: 16339467
- National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke (NINDS trial). N Engl J Med. 1995;333(24):1581-1587. PMID: 7477192
- Hacke W, Kaste M, Bluhmki E, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke (ECASS III). N Engl J Med. 2008;359(13):1317-1329. PMID: 18815396
- Campbell BCV, Mitchell PJ, Churilov L, et al. Tenecteplase versus alteplase before thrombectomy for ischemic stroke (EXTEND-IA TNK). N Engl J Med. 2018;378(17):1573-1582. PMID: 29766771
- Berkhemer OA, Fransen PS, Beumer D, et al. A randomized trial of intraarterial treatment for acute ischemic stroke (MR CLEAN). N Engl J Med. 2015;372(1):11-20. PMID: 25517348
- Nogueira RG, Jadhav AP, Haussen DC, et al. Thrombectomy 6 to 24 hours after stroke with a mismatch between deficit and infarct (DAWN trial). N Engl J Med. 2018;378(1):11-21. PMID: 29364767
- Albers GW, Marks MP, Kemp S, et al. Thrombectomy for stroke at 6 to 16 hours with selection by perfusion imaging (DEFUSE 3). N Engl J Med. 2018;378(8):708-718. PMID: 29414764
- Johnston SC, Easton JD, Farrant M, et al. Clopidogrel and aspirin in acute ischemic stroke and high-risk TIA (POINT trial). N Engl J Med. 2018;379(3):215-225. PMID: 29766750
- Anderson CS, Heeley E, Huang Y, et al. Rapid blood-pressure lowering in patients with acute intracerebral hemorrhage (INTERACT2). N Engl J Med. 2013;368(25):2355-2365. PMID: 23713578
- Molyneux AJ, Kerr RS, Yu LM, et al. International Subarachnoid Aneurysm Trial (ISAT) of neurosurgical clipping versus endovascular coiling. Lancet. 2005;366(9488):809-817. PMID: 16360683
- Silbergleit R, Durkalski V, Lowenstein D, et al. Intramuscular versus intravenous therapy for prehospital status epilepticus (RAMPART). N Engl J Med. 2012;366(7):591-600. PMID: 22335736
- Kapur J, Elm J, Chamberlain JM, et al. Randomized trial of three anticonvulsant medications for status epilepticus (ESETT). N Engl J Med. 2019;381(22):2103-2113. PMID: 31774955
- Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the ILAE (2017). Epilepsia. 2017;58(4):522-530. PMID: 28276060
Textbooks & Reference Works
- Adams and Victor's Principles of Neurology. 12th ed. Ropper AH, Samuels MA, Klein JP, Prasad S, eds. McGraw-Hill; 2023.
- Bradley and Daroff's Neurology in Clinical Practice. 8th ed. Jankovic J, Mazziotta JC, Pomeroy SL, Newman NJ, eds. Elsevier; 2022.
- Gray's Anatomy: The Anatomical Basis of Clinical Practice. 42nd ed. Standring S, ed. Elsevier; 2020.