Rheumatology
Every autoimmune disease, crystal arthropathy, vasculitis, connective tissue disorder, classification criterion, autoantibody, DMARD, biologic, and management strategy in one place.
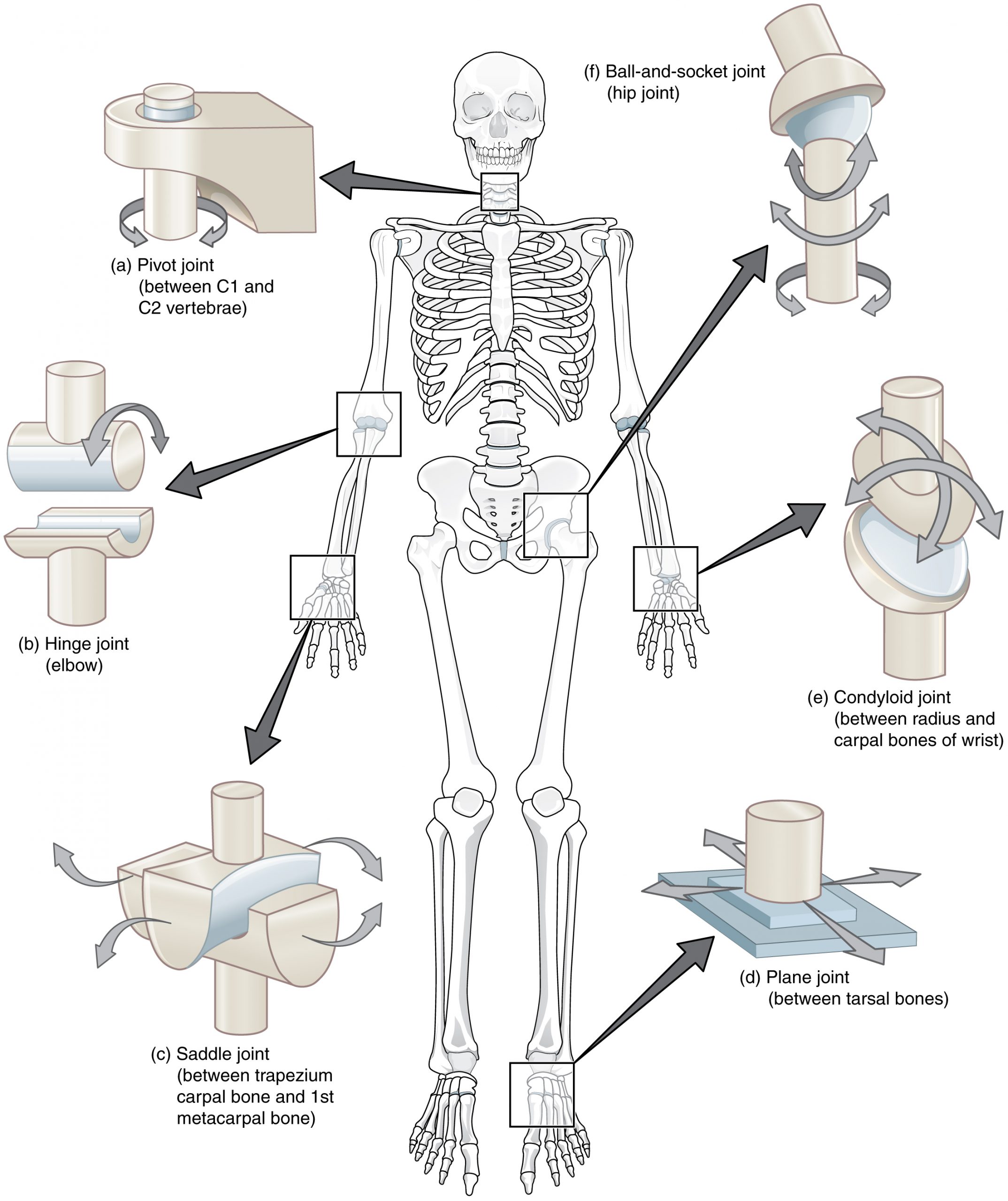
01 Musculoskeletal & Immunologic Anatomy
Rheumatology encompasses diseases of the joints, connective tissues, and immune system. A solid understanding of joint architecture, the synovial membrane, articular cartilage, and the innate and adaptive immune responses is essential for understanding every disease in this specialty.
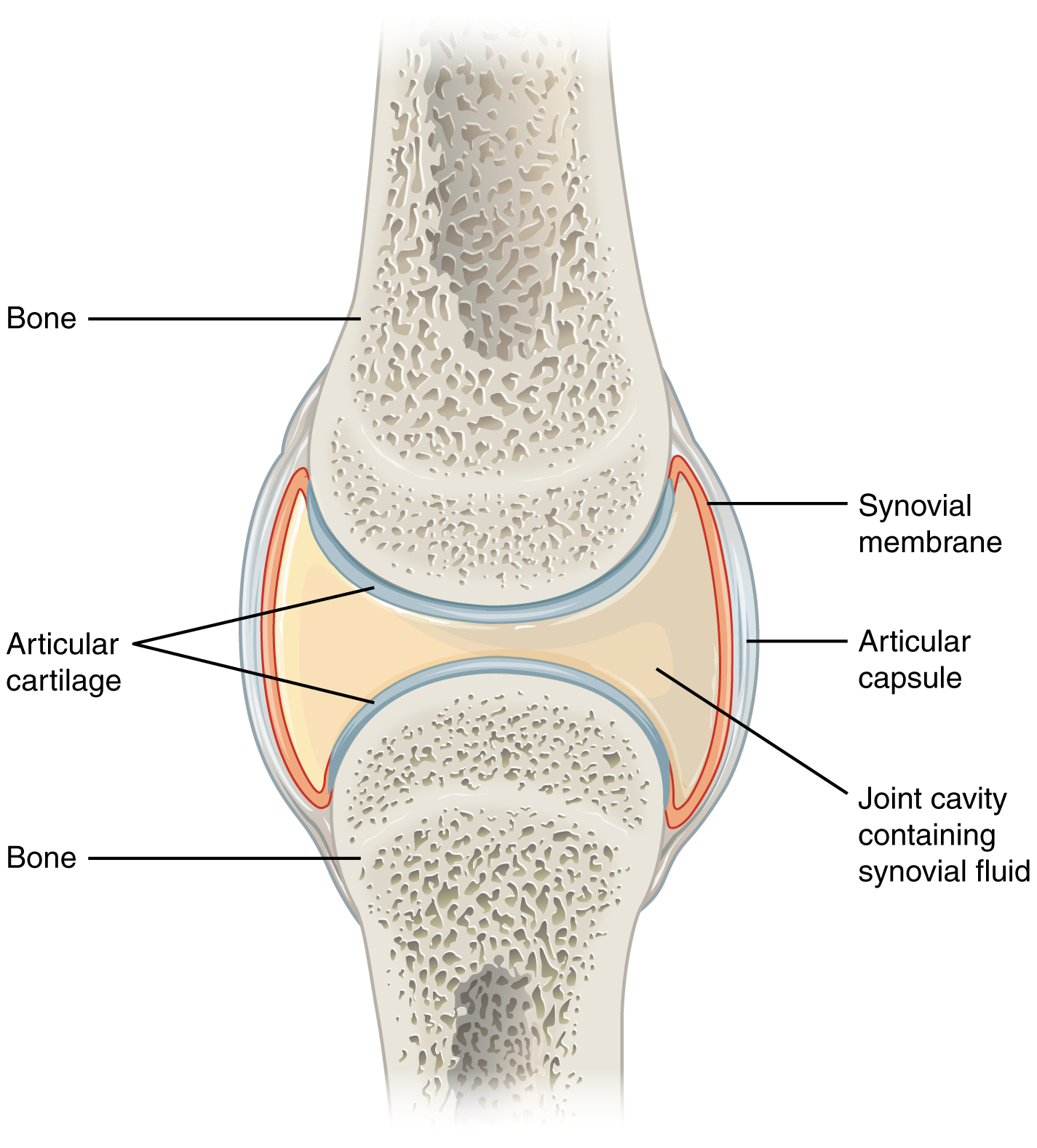
Synovial Joint Structure
A synovial (diarthrodial) joint is the most common and most mobile joint type. It consists of two bone ends covered with hyaline articular cartilage (2–4 mm thick, avascular, composed of type II collagen and proteoglycans in a water-rich matrix), enclosed within a joint capsule. The capsule has two layers: an outer fibrous layer (dense connective tissue providing mechanical stability) and an inner synovial membrane (synovium). The synovium is 1–3 cells thick, composed of type A synoviocytes (macrophage-like, phagocytic) and type B synoviocytes (fibroblast-like, producing hyaluronic acid and lubricin). Normal synovial fluid is clear, viscous, and contains <200 WBC/mm³; it provides lubrication and nutrient delivery to the avascular cartilage.
Cartilage Biology
Chondrocytes are the sole cell type in articular cartilage, maintaining the extracellular matrix (ECM) through balanced synthesis and degradation of type II collagen and aggrecan (the major proteoglycan). In osteoarthritis, matrix metalloproteinases (MMPs) and aggrecanases (ADAMTS-4/5) overwhelm synthetic capacity, leading to progressive cartilage loss. In inflammatory arthritis (e.g., RA), cytokines such as TNF-alpha, IL-1, and IL-6 from the inflamed synovium drive MMP production and inhibit chondrocyte repair, causing erosive joint destruction.
The Innate Immune System
The innate immune system provides immediate, nonspecific defense. Key components include: neutrophils (the dominant cell in septic and crystal arthritis synovial fluid), macrophages (antigen presentation, cytokine production — TNF-alpha, IL-1, IL-6), natural killer (NK) cells, the complement system (classical, alternative, lectin pathways), and pattern recognition receptors (toll-like receptors, NOD-like receptors). The NLRP3 inflammasome is critical in crystal arthritis — monosodium urate (MSU) crystals activate NLRP3, leading to caspase-1 activation, IL-1-beta release, and the acute gout flare.
The Adaptive Immune System
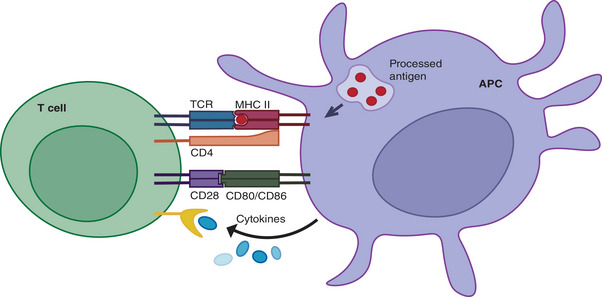
T lymphocytes mature in the thymus and are divided into CD4+ helper T cells (Th1, Th2, Th17, Treg subtypes) and CD8+ cytotoxic T cells. Th17 cells produce IL-17 and are central to spondyloarthropathy pathogenesis. Regulatory T cells (Tregs) suppress autoimmune responses; their dysfunction contributes to loss of self-tolerance. B lymphocytes mature in the bone marrow, produce antibodies (immunoglobulins), and serve as antigen-presenting cells. Autoantibodies (RF, anti-CCP, ANA, ANCA) are the hallmark of many rheumatic diseases. B-cell depletion with rituximab (anti-CD20) is effective in RA, ANCA vasculitis, and other conditions. HLA molecules (MHC class I and II) present antigens to T cells; specific HLA alleles confer disease susceptibility — e.g., HLA-B27 (ankylosing spondylitis), HLA-DR4 (RA), HLA-DR2/DR3 (SLE).
The Complement System
Three activation pathways converge on C3 convertase:
| Pathway | Trigger | Key Components | Clinical Relevance |
|---|---|---|---|
| Classical | Antigen-antibody complexes (IgG, IgM) | C1q, C1r, C1s → C4, C2 | SLE (C1q, C2, C4 deficiency predisposes to SLE) |
| Alternative | Spontaneous C3 hydrolysis, microbial surfaces | Factor B, Factor D, properdin | Complement-mediated aHUS |
| Lectin | Mannose on microbial surfaces | MBL, MASP-1/2 | Recurrent infections if MBL deficient |
All three pathways generate C3a/C5a (anaphylatoxins causing inflammation) and the membrane attack complex (MAC, C5b-9) causing cell lysis. In SLE, immune complex deposition activates the classical pathway, consuming C3 and C4 — low complement levels correlate with disease activity, especially lupus nephritis.
Autoimmunity — Loss of Self-Tolerance
Autoimmune disease results from failure of central tolerance (thymic deletion of self-reactive T cells, bone marrow deletion of self-reactive B cells) and/or peripheral tolerance (Tregs, anergy, peripheral deletion). Contributing factors include: genetic susceptibility (HLA associations, PTPN22, STAT4), environmental triggers (infections via molecular mimicry, smoking in RA/anti-CCP, UV light in SLE, silica in scleroderma), epigenetic modifications (DNA hypomethylation in SLE), and hormonal factors (female predominance in most autoimmune diseases — estrogen enhances B-cell survival and antibody production).
02 The Rheumatologic Exam
The rheumatologic physical examination is a systematic assessment of joints, periarticular structures, skin, nails, and vascular status. It is the single most important skill in rheumatology — laboratory tests and imaging are adjuncts to, not substitutes for, a thorough exam.
The Joint Count
The 28-joint count (used in DAS28) assesses bilateral MCPs (1–5), PIPs (1–5), wrists, elbows, shoulders, and knees for tenderness (reported by patient on palpation) and swelling (detected by examiner — boggy soft-tissue thickening over the joint line, distinct from bony enlargement). The 66/68 joint count adds feet, ankles, hips, and additional hand joints. Document each joint as tender and/or swollen separately.
Swelling vs. Tenderness
| Finding | Significance | Assessment |
|---|---|---|
| Synovial swelling (boggy) | Active synovitis — hallmark of inflammatory arthritis | Palpate over joint line; fluctuant soft-tissue fullness |
| Bony enlargement | OA (Heberden/Bouchard nodes); chronic remodeling | Hard, non-tender bony prominences at DIP/PIP |
| Effusion | Intra-articular fluid; inflammatory or mechanical | Bulge sign (knee), ballottement of patella |
| Tenderness without swelling | Enthesitis, fibromyalgia, tendinopathy | Localize to joint line vs. periarticular structures |
The Schober Test
Assesses lumbar spine flexion in suspected ankylosing spondylitis. With the patient standing erect, mark the skin at the level of the posterior superior iliac spines (approximately L5) and 10 cm above. Ask the patient to flex forward maximally. Normal: the distance between marks increases by ≥5 cm (to ≥15 cm). An increase of <5 cm suggests restricted lumbar flexion and is characteristic of axial spondyloarthropathy.
Hand Examination
| Finding | Location | Disease Association |
|---|---|---|
| Heberden nodes | DIP joints | Osteoarthritis |
| Bouchard nodes | PIP joints | Osteoarthritis |
| Swan-neck deformity | PIP hyperextension + DIP flexion | Rheumatoid arthritis, SLE |
| Boutonniere deformity | PIP flexion + DIP hyperextension | Rheumatoid arthritis |
| Ulnar deviation | MCPs deviate ulnarly | Rheumatoid arthritis |
| Z-thumb deformity | CMC hyperextension + MCP flexion | Rheumatoid arthritis |
| Dactylitis ("sausage digit") | Entire digit swollen | Psoriatic arthritis, reactive arthritis |
| Telescoping digits | Shortened fingers with redundant skin | Psoriatic arthritis (arthritis mutilans) |
| Gottron papules | MCPs, PIPs, DIPs (extensor surfaces) | Dermatomyositis |
| Mechanic's hands | Cracked, fissured skin on fingers/palms | Anti-synthetase syndrome |
| Sclerodactyly | Taut, shiny, thickened skin over digits | Systemic sclerosis |
Nail Examination
Nail pitting (small depressions in nail plate) is seen in up to 80% of psoriatic arthritis. Other nail changes: onycholysis (separation from nail bed), subungual hyperkeratosis, oil-drop discoloration (yellow-brown patches), and nail-fold capillary changes (dilated loops in scleroderma/dermatomyositis, seen with dermatoscopy or ophthalmoscope). Splinter hemorrhages may indicate vasculitis or endocarditis.
Skin Thickening Assessment (Modified Rodnan Skin Score)
Used in systemic sclerosis. Palpate skin at 17 body sites (fingers, hands, forearms, upper arms, face, chest, abdomen, thighs, lower legs, feet). Score each site 0–3 (0 = normal; 1 = mild thickening; 2 = moderate, cannot pinch; 3 = severe, unable to move skin). Total score 0–51. Scores >20 indicate severe diffuse disease; serial measurements track treatment response.
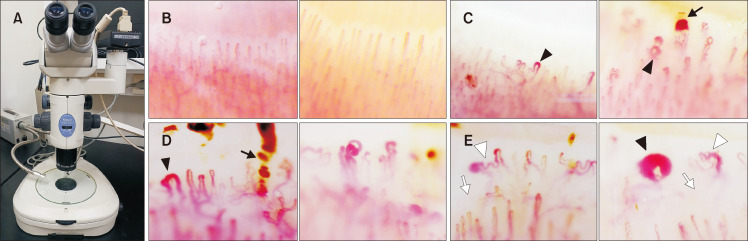
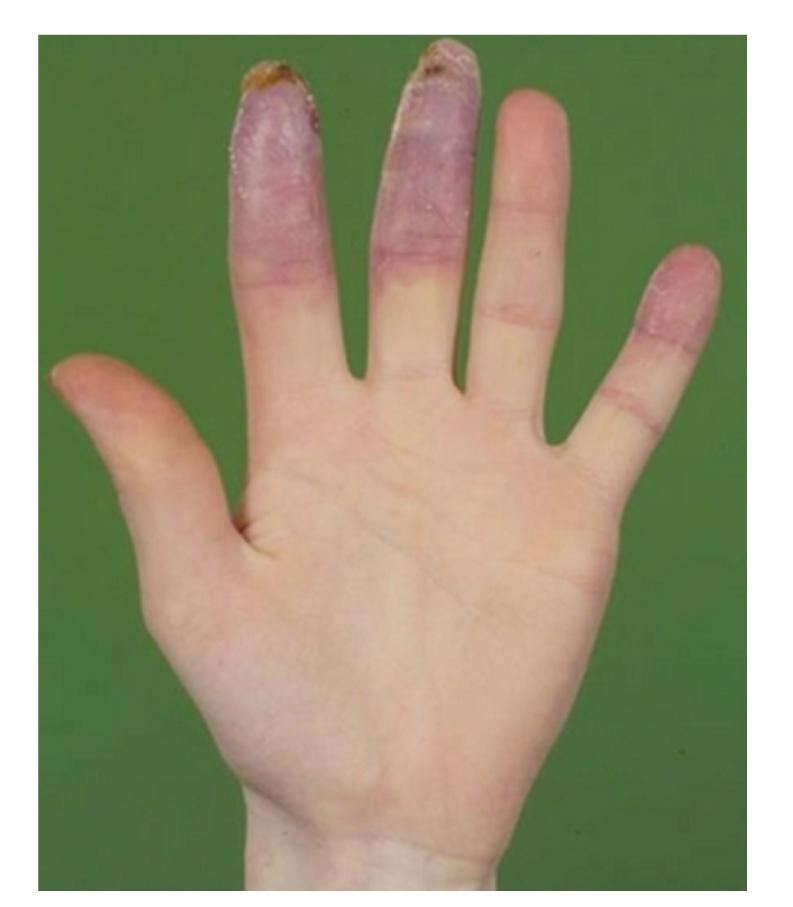
Raynaud Phenomenon Assessment
Triphasic color change: white (vasospasm/pallor) → blue (cyanosis/deoxygenation) → red (reperfusion hyperemia). Primary Raynaud (80–90% of cases) is symmetric, mild, without tissue damage, ANA-negative, and normal nail-fold capillaries. Secondary Raynaud (associated with CTD, especially scleroderma) features asymmetric attacks, digital ulcers/pitting scars, abnormal nail-fold capillaries, and positive ANA.
03 Key Terminology & Abbreviations
Rheumatology uses an extensive set of abbreviations for diseases, autoantibodies, medications, and classification systems. Familiarity with these terms is essential for reading clinical documentation and understanding treatment guidelines.
| Abbreviation | Meaning |
|---|---|
| RA | Rheumatoid arthritis |
| SLE | Systemic lupus erythematosus |
| OA | Osteoarthritis |
| SpA | Spondyloarthropathy |
| AS | Ankylosing spondylitis |
| PsA | Psoriatic arthritis |
| SSc | Systemic sclerosis (scleroderma) |
| DM / PM | Dermatomyositis / polymyositis |
| SS | Sjögren syndrome |
| MCTD | Mixed connective tissue disease |
| GCA | Giant cell arteritis (temporal arteritis) |
| GPA | Granulomatosis with polyangiitis (formerly Wegener) |
| MPA | Microscopic polyangiitis |
| EGPA | Eosinophilic granulomatosis with polyangiitis (formerly Churg-Strauss) |
| PAN | Polyarteritis nodosa |
| ANA | Antinuclear antibody |
| RF | Rheumatoid factor |
| Anti-CCP | Anti-cyclic citrullinated peptide antibody |
| dsDNA | Anti-double-stranded DNA antibody |
| ENA | Extractable nuclear antigens (anti-Sm, anti-RNP, SSA, SSB) |
| ANCA | Anti-neutrophil cytoplasmic antibody (c-ANCA / p-ANCA) |
| DMARD | Disease-modifying antirheumatic drug |
| csDMARD | Conventional synthetic DMARD (MTX, HCQ, SSZ, LEF) |
| bDMARD | Biologic DMARD (TNFi, IL-6i, rituximab, abatacept) |
| tsDMARD | Targeted synthetic DMARD (JAK inhibitors) |
| MTX | Methotrexate |
| HCQ | Hydroxychloroquine |
| SSZ | Sulfasalazine |
| LEF | Leflunomide |
| MMF | Mycophenolate mofetil |
| AZA | Azathioprine |
| CYC | Cyclophosphamide |
| TNFi | TNF inhibitor (etanercept, infliximab, adalimumab, golimumab, certolizumab) |
| ULT | Urate-lowering therapy |
| DAS28 | Disease Activity Score (28 joints) |
| CDAI | Clinical Disease Activity Index |
| SLEDAI | SLE Disease Activity Index |
| BVAS | Birmingham Vasculitis Activity Score |
04 RA Pathophysiology & Diagnosis
Rheumatoid arthritis (RA) is a chronic, systemic autoimmune disease characterized by symmetric inflammatory polyarthritis that, if untreated, leads to progressive joint destruction. It affects ~1% of the global population, with a female-to-male ratio of 3:1, and peak onset between ages 30–60.
Pathophysiology
RA begins with loss of tolerance to citrullinated self-proteins. Citrullination (conversion of arginine to citrulline by peptidylarginine deiminase [PAD] enzymes) occurs normally but is enhanced by smoking, periodontitis (Porphyromonas gingivalis produces PAD), and mucosal inflammation. Genetically susceptible individuals (HLA-DR4 shared epitope) generate anti-citrullinated protein antibodies (ACPA/anti-CCP) and rheumatoid factor (RF) years before clinical disease. In the joint, immune complex deposition and T-cell activation drive synovial inflammation with formation of pannus — an aggressive, tumor-like synovial tissue that invades cartilage and bone via osteoclast activation (RANKL pathway) and MMP/cytokine release (TNF-alpha, IL-1, IL-6).
2010 ACR/EULAR Classification Criteria
A score ≥6/10 classifies definite RA (requires at least 1 clinically swollen joint not better explained by another disease):
| Domain | Category | Score |
|---|---|---|
| Joint involvement | 1 large joint | 0 |
| 2–10 large joints | 1 | |
| 1–3 small joints (± large joints) | 2 | |
| 4–10 small joints (± large joints) | 3 | |
| >10 joints (at least 1 small joint) | 5 | |
| Serology | Negative RF AND negative anti-CCP | 0 |
| Low-positive RF OR low-positive anti-CCP | 2 | |
| High-positive RF OR high-positive anti-CCP (>3× ULN) | 3 | |
| Acute phase reactants | Normal CRP AND normal ESR | 0 |
| Abnormal CRP OR abnormal ESR | 1 | |
| Duration of symptoms | <6 weeks | 0 |
| ≥6 weeks | 1 |
Laboratory Findings
| Test | Sensitivity | Specificity | Notes |
|---|---|---|---|
| Rheumatoid Factor (RF) | 60–80% | ~80% | IgM against Fc portion of IgG; also positive in infections, other CTDs, elderly |
| Anti-CCP (ACPA) | 60–75% | 95–99% | More specific than RF; predicts erosive disease; can precede symptoms by years |
| ESR / CRP | Variable | Nonspecific | Track disease activity; CRP responds faster than ESR |
Radiographic Findings
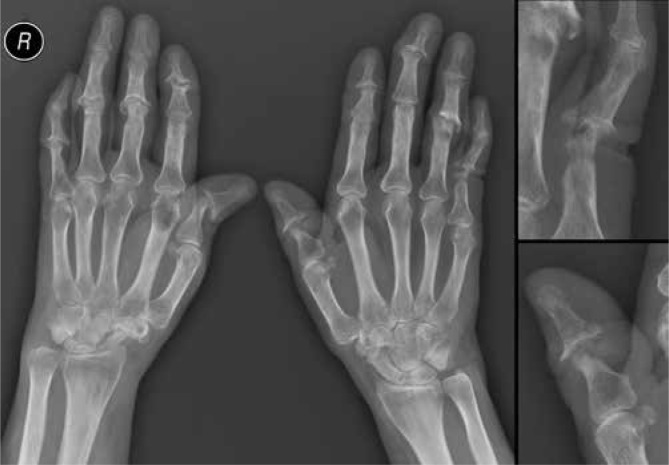
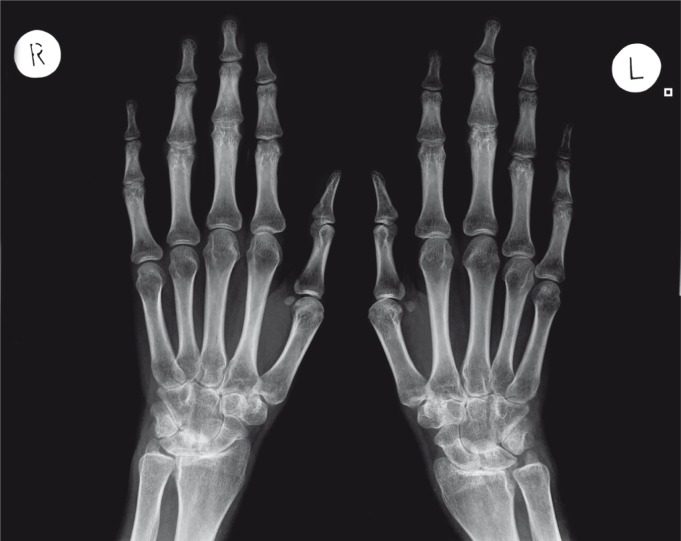
Early RA: periarticular soft-tissue swelling, juxta-articular osteopenia. Progressive: symmetric joint space narrowing (loss of cartilage), marginal erosions (at bare areas where bone is not covered by cartilage). Late: subluxation, ankylosis. Classic locations: MCP, PIP, wrists, MTP joints. DIP involvement suggests OA or PsA, NOT RA.
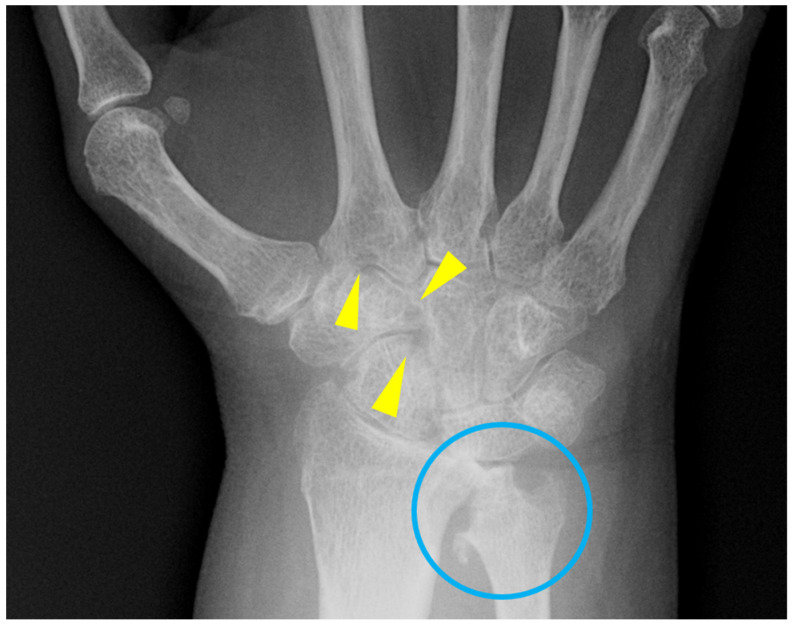
05 RA Treatment
The modern RA treatment paradigm is treat-to-target (T2T): aim for remission (or low disease activity if remission is not achievable) with frequent monitoring and rapid escalation of therapy.
Treat-to-Target Approach
| Step | Intervention | Timeline |
|---|---|---|
| 1 | Start methotrexate (MTX) 15–25 mg/week (oral or SC) + folic acid 1 mg/day | At diagnosis |
| 2 | Reassess disease activity (DAS28, CDAI) every 1–3 months | Ongoing |
| 3 | If target not met in 3 months: optimize MTX dose (25 mg SC) or add/switch csDMARD | 3 months |
| 4 | If target not met at 6 months despite csDMARDs: add bDMARD or tsDMARD | 6 months |
| 5 | If first bDMARD fails: switch mechanism of action | 3–6 months after bDMARD |
Conventional Synthetic DMARDs (csDMARDs)
| Drug | Dose | Mechanism | Key Monitoring | Adverse Effects |
|---|---|---|---|---|
| Methotrexate (MTX) | 15–25 mg PO/SC weekly | Folate antagonist; inhibits AICAR transformylase → adenosine release | CBC, LFTs, creatinine q8–12 weeks | Hepatotoxicity, cytopenias, stomatitis, pneumonitis; teratogenic |
| Hydroxychloroquine (HCQ) | 200–400 mg PO daily (≤5 mg/kg/day) | Inhibits TLR signaling, antigen processing | Baseline and annual ophthalmology after 5 years (retinal toxicity) | Retinal toxicity, GI upset, QTc prolongation (rare) |
| Sulfasalazine (SSZ) | 1–3 g PO daily (divided) | Anti-inflammatory; inhibits NF-kB | CBC, LFTs q8–12 weeks | GI upset, rash, cytopenias, oligospermia (reversible) |
| Leflunomide (LEF) | 20 mg PO daily (loading: 100 mg x3 days optional) | Pyrimidine synthesis inhibitor (DHODH) | CBC, LFTs monthly x6 months then q8 weeks | Hepatotoxicity, diarrhea, alopecia; teratogenic (long washout — cholestyramine needed) |
Biologic DMARDs (bDMARDs)
| Target | Agents | Route | Key Considerations |
|---|---|---|---|
| TNF-alpha | Etanercept, infliximab, adalimumab, golimumab, certolizumab | SC or IV | First-line biologics; screen for TB/hepatitis B before starting; avoid in active infection, heart failure NYHA III-IV, demyelinating disease |
| IL-6 receptor | Tocilizumab, sarilumab | SC or IV | CRP may be suppressed (unreliable marker); monitor lipids; risk of GI perforation (diverticulitis history) |
| T-cell co-stimulation (CD80/86) | Abatacept | SC or IV | Favorable safety profile; may be preferred in patients with COPD or recurrent infections |
| CD20 (B-cell depletion) | Rituximab | IV (1000 mg x2 doses, 2 weeks apart, q6 months) | Preferred in RF/anti-CCP positive; screen for hepatitis B; check immunoglobulin levels |
Targeted Synthetic DMARDs (tsDMARDs) — JAK Inhibitors
| Drug | JAK Selectivity | Dose | Key Safety Concerns |
|---|---|---|---|
| Tofacitinib | JAK1/JAK3 | 5 mg PO BID | Increased risk of VTE, MACE, malignancy (especially in patients >65, smokers, cardiovascular risk factors) per FDA boxed warning; herpes zoster reactivation; cytopenias |
| Baricitinib | JAK1/JAK2 | 2 mg PO daily | |
| Upadacitinib | JAK1-selective | 15 mg PO daily |
Bridging Therapy
Glucocorticoids (prednisone 5–10 mg/day or equivalent) provide rapid symptom relief while DMARDs take effect (MTX onset: 4–8 weeks). Taper and discontinue within 3–6 months. Long-term low-dose steroids increase infection, osteoporosis, and cardiovascular risk. Intra-articular steroid injections are useful for persistent monoarthritis.
06 RA Complications
Articular Complications
| Complication | Mechanism | Clinical Features | Management |
|---|---|---|---|
| Atlantoaxial subluxation | Pannus erodes the transverse ligament of C1 | Neck pain, occipital headache; risk of cord compression (myelopathy, quadriplegia) | Flexion/extension C-spine X-ray or MRI; surgical fixation if subluxation >9 mm or myelopathy |
| Joint destruction/deformity | Pannus invasion of cartilage and bone | Swan-neck, boutonniere, ulnar deviation, Z-thumb | Early aggressive DMARD therapy; joint replacement if severe |
| Baker cyst (popliteal cyst) | Synovial fluid accumulation posteriorly | Posterior knee swelling; rupture mimics DVT | Treat underlying synovitis; aspiration; ultrasound to distinguish from DVT |
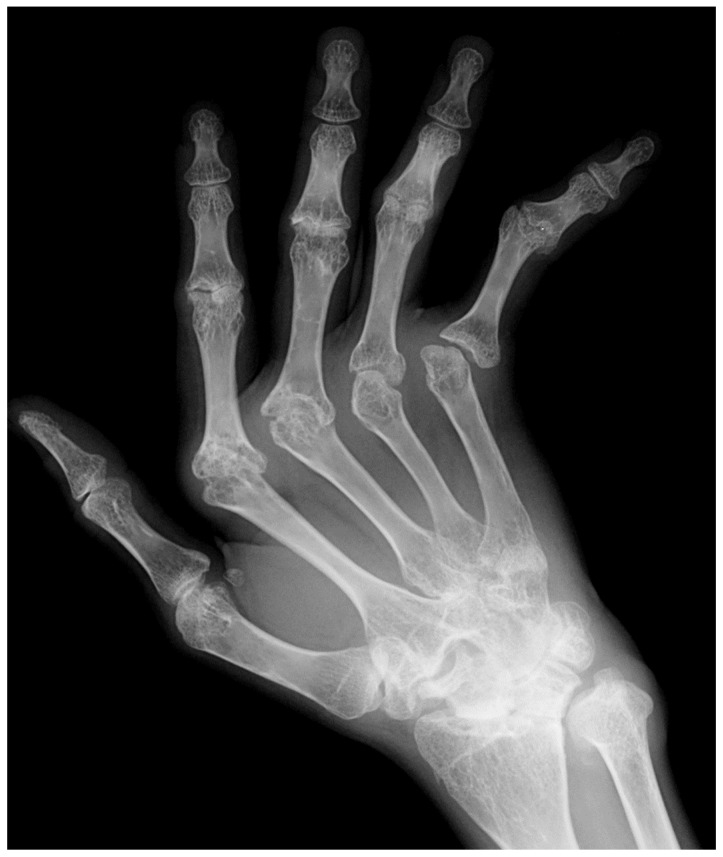
Extra-Articular Manifestations
| System | Manifestation | Details |
|---|---|---|
| Pulmonary | Interstitial lung disease (ILD) | Most common cause of RA-related death after CV disease; UIP or NSIP pattern; screen high-risk patients with PFTs/HRCT |
| Pulmonary | Pleural effusions | Exudative, very low glucose (<30 mg/dL), high LDH, low complement |
| Pulmonary | Rheumatoid nodules (pulmonary) | Single or multiple; Caplan syndrome = nodules + coal workers' pneumoconiosis |
| Hematologic | Felty syndrome | RA + splenomegaly + neutropenia; high RF titers; increased infection risk |
| Hematologic | Large granular lymphocyte (LGL) leukemia | Associated with RA; clonal T-cell expansion; causes neutropenia similar to Felty |
| Skin | Rheumatoid nodules | Subcutaneous nodules over extensor surfaces (olecranon, fingers); occur in 20–30% of seropositive RA |
| Cardiac | Pericarditis | Most common cardiac manifestation; usually subclinical |
| Ocular | Scleritis, episcleritis, keratoconjunctivitis sicca | Scleritis can be necrotizing (scleromalacia perforans) — vision-threatening |
| Vascular | Rheumatoid vasculitis | Rare, severe; digital infarcts, skin ulcers, neuropathy; associated with high-titer RF, nodular disease |
| Cardiovascular | Accelerated atherosclerosis | RA is an independent CV risk factor; treat inflammation aggressively; lipid management |
07 SLE Pathophysiology & Diagnosis
Systemic lupus erythematosus (SLE) is the prototypical systemic autoimmune disease, characterized by autoantibody production, immune complex deposition, and multi-organ inflammation. It affects women of childbearing age preferentially (F:M = 9:1), with higher incidence and severity in Black, Hispanic, and Asian populations.
Pathophysiology
SLE results from defective clearance of apoptotic cells and nuclear debris, leading to exposure of intracellular antigens (dsDNA, histones, Sm, RNP) to the immune system. Key mechanisms include: (1) Type III hypersensitivity — immune complex deposition in kidneys, skin, joints, serosal surfaces with complement activation; (2) Type II hypersensitivity — autoantibodies against blood cells causing hemolytic anemia, thrombocytopenia, leukopenia; (3) Interferon-alpha pathway — plasmacytoid dendritic cells produce excessive IFN-alpha in response to nucleic acid-containing immune complexes via TLR7/9, driving B-cell activation and autoantibody production.
2019 EULAR/ACR Classification Criteria
Entry criterion: ANA ≥1:80 (or equivalent). Then apply additive criteria (do not count if better explained by another diagnosis). Classify as SLE if total score ≥10:
| Domain | Criterion | Score |
|---|---|---|
| Constitutional | Fever (>38.3°C) | 2 |
| Hematologic | ||
| Leukopenia (<4000/μL) | 3 | |
| Thrombocytopenia (<100,000/μL) | 4 | |
| Autoimmune hemolysis | 4 | |
| Neuropsychiatric | Delirium | 2 |
| Psychosis | 3 | |
| Seizure | 5 | |
| Mucocutaneous | Non-scarring alopecia | 2 |
| Oral ulcers | 2 | |
| Subacute cutaneous OR discoid lupus | 4 | |
| Acute cutaneous lupus (malar rash) | 6 | |
| Serosal | Pleural or pericardial effusion | 5 |
| Acute pericarditis | 6 | |
| Musculoskeletal | Joint involvement (≥2 joints with synovitis OR tenderness + morning stiffness ≥30 min) | 6 |
| Renal | Proteinuria >0.5 g/24h | 4 |
| Class II or V lupus nephritis on biopsy | 8 | |
| Class III or IV lupus nephritis on biopsy | 10 | |
| Immunologic | ||
| Anti-dsDNA OR anti-Sm | 6 | |
| Low C3 OR low C4 | 3 | |
| Low C3 AND low C4 | 4 | |
| Antiphospholipid antibodies (aCL, anti-B2GP1, or lupus anticoagulant) | 2 | |
ANA Patterns and Associations
| ANA Pattern | Target Antigen | Disease Association |
|---|---|---|
| Homogeneous (diffuse) | dsDNA, histones | SLE, drug-induced lupus |
| Speckled | Sm, RNP, SSA, SSB | SLE, MCTD, Sjögren |
| Nucleolar | RNA polymerase III, Scl-70, fibrillarin | Systemic sclerosis |
| Centromere | CENP-A, B, C | Limited systemic sclerosis (CREST) |
| Cytoplasmic | Jo-1, ribosomal P | Myositis (anti-synthetase), SLE |
08 SLE Organ Manifestations
Mucocutaneous
| Manifestation | Description | Key Features |
|---|---|---|
| Acute cutaneous lupus (malar rash) | Erythematous, flat or raised rash over cheeks and nasal bridge | Spares nasolabial folds; photosensitive; may be transient |
| Subacute cutaneous lupus (SCLE) | Annular polycyclic or papulosquamous lesions | Strongly associated with anti-SSA/Ro; photosensitive; heals without scarring |
| Discoid lupus | Erythematous plaques with adherent scale, follicular plugging | Scarring alopecia; can occur without systemic SLE (~5% progress to SLE) |
| Oral ulcers | Usually painless; hard palate most specific location | Often overlooked; ask specifically |
| Non-scarring alopecia | Diffuse hair thinning | Lupus hair — fragile, broken hairs at the hairline |
Musculoskeletal
Jaccoud arthropathy is the classic lupus joint disease — non-erosive, reducible deformities (ulnar deviation, swan-neck) caused by ligament and tendon laxity, NOT bony erosion (distinguishes from RA). Arthritis occurs in ~90% of SLE patients and is typically symmetric, involving small joints. Osteonecrosis (avascular necrosis) of the femoral head occurs in ~5–10% of SLE patients, often related to corticosteroid use.
Serositis
Pleuritis (most common serosal manifestation, ~50%) and pericarditis (~25%) are typical. Lupus pleuritis produces exudative effusions with low complement. Shrinking lung syndrome (unexplained dyspnea with reduced lung volumes and elevated diaphragms without parenchymal disease) is a rare but characteristic manifestation.
Hematologic
| Cytopenia | Mechanism | Clinical Notes |
|---|---|---|
| Leukopenia / lymphopenia | Anti-lymphocyte antibodies; active disease marker | Lymphopenia <1000/μL is most common; correlates with disease activity |
| Hemolytic anemia | Warm autoimmune hemolytic anemia (IgG, positive DAT) | Reticulocytosis, elevated LDH, low haptoglobin, indirect bilirubinemia |
| Thrombocytopenia | Anti-platelet antibodies; may precede SLE diagnosis by years | Consider ITP vs. TTP vs. APS-associated thrombocytopenia |
Neuropsychiatric Lupus (NPSLE)
Occurs in 30–40% of SLE patients. The most common manifestation is cognitive dysfunction ("lupus fog"). Severe manifestations include seizures, psychosis, transverse myelitis, cerebrovascular disease (often APS-related), and cranial neuropathies. Diagnosis is one of exclusion (rule out infection, metabolic, drug effects). Anti-ribosomal P antibodies are associated with lupus psychosis. CSF may show elevated protein, mild pleocytosis. MRI may show white matter lesions.
Cardiovascular
Libman-Sacks endocarditis — sterile verrucous vegetations on the undersurface of valve leaflets (typically mitral), associated with antiphospholipid antibodies. Accelerated atherosclerosis is a leading cause of late mortality in SLE. Myocarditis is uncommon but serious.
09 Lupus Nephritis
Lupus nephritis (LN) is the most important organ manifestation of SLE, occurring in 40–60% of patients. It is a major determinant of morbidity and mortality. All SLE patients should be screened with urinalysis and serum creatinine at every visit.
ISN/RPS Classification (2003, revised 2018)
| Class | Pattern | Description | Prognosis / Treatment |
|---|---|---|---|
| I | Minimal mesangial | Normal light microscopy; mesangial immune deposits on IF/EM only | Excellent; no specific treatment needed |
| II | Mesangial proliferative | Mesangial hypercellularity with immune deposits | Good; usually HCQ alone or low-dose immunosuppression |
| III | Focal proliferative | <50% of glomeruli affected; active/chronic endocapillary or extracapillary lesions | Requires induction immunosuppression |
| IV | Diffuse proliferative | ≥50% of glomeruli affected; most common and most severe | Most aggressive treatment required; highest risk of ESRD |
| V | Membranous | Subepithelial immune deposits; nephrotic syndrome | Moderate prognosis; treat if proteinuria >1 g/day despite RAAS blockade |
| VI | Advanced sclerosing | ≥90% globally sclerosed glomeruli; end-stage | Irreversible; prepare for RRT |
Biopsy Indications
Renal biopsy is indicated in SLE patients with: proteinuria >0.5 g/24h (or protein/creatinine ratio >0.5), active urine sediment (RBC casts, dysmorphic RBCs), unexplained rising creatinine, or failure to respond to treatment. Biopsy results guide treatment intensity.
Treatment by Class
| Phase | Class III/IV | Class V (Pure Membranous) |
|---|---|---|
| Induction (6 months) | Mycophenolate mofetil (MMF) 2–3 g/day (preferred, especially in Black/Hispanic patients) OR IV cyclophosphamide (Euro-Lupus low-dose protocol: 500 mg q2 weeks × 6 doses) | MMF 2–3 g/day OR calcineurin inhibitor (tacrolimus/voclosporin) |
| Maintenance (3–5+ years) | MMF 1–2 g/day (preferred) OR azathioprine 2 mg/kg/day | MMF 1–2 g/day |
| Adjuncts | HCQ (ALL patients), ACEi/ARB for proteinuria, prednisone taper (start 0.5–1 mg/kg, taper to ≤5 mg by 3–6 months), belimumab (anti-BLyS, FDA-approved for active LN), voclosporin (calcineurin inhibitor, FDA-approved for LN in combination with MMF) | |
10 SLE Treatment
Hydroxychloroquine — The Foundation
HCQ should be prescribed to EVERY SLE patient unless contraindicated. Benefits include: reduced flares by 50%, reduced organ damage accrual, reduced thrombosis risk (especially in APS), improved lipid profiles, reduced infection risk, improved survival, and protection against lupus nephritis flares. Dose: ≤5 mg/kg actual body weight/day. Monitor with annual OCT (optical coherence tomography) after 5 years or sooner if risk factors (renal impairment, tamoxifen use, high dose).
Treatment by Disease Severity
| Severity | Manifestations | Treatment |
|---|---|---|
| Mild | Skin, arthritis, fatigue, serositis | HCQ + low-dose prednisone (≤7.5 mg/day) + NSAIDs; belimumab if persistent; consider MTX for arthritis |
| Moderate | Refractory skin/joints, hematologic (moderate cytopenias) | HCQ + MMF or AZA + short-course steroids; anifrolumab (anti-IFNAR1) for skin/joint predominant disease |
| Severe | Nephritis (III/IV/V), CNS lupus, alveolar hemorrhage, severe cytopenias | HCQ + high-dose pulse steroids (methylprednisolone 500–1000 mg IV x3 days) + MMF or CYC; rituximab for refractory; belimumab or voclosporin add-on for LN |
| Life-threatening | Diffuse alveolar hemorrhage, cerebritis, TTP | Pulse steroids + CYC or rituximab; plasma exchange for TTP; IVIG for severe thrombocytopenia |
Newer Therapies
| Drug | Target | Indication | Key Data |
|---|---|---|---|
| Belimumab | BLyS (BAFF) | Active SLE; active lupus nephritis | BLISS-LN: improved renal response + reduced renal flares; first biologic approved for SLE |
| Anifrolumab | Type I IFN receptor (IFNAR1) | Active SLE (skin and joint predominant) | TULIP trials: improved BICLA response; most benefit in IFN-high patients |
| Voclosporin | Calcineurin (calcineurin inhibitor) | Active lupus nephritis (with MMF) | AURORA: 41% complete renal response vs. 23% placebo at 52 weeks |
11 Gout
Gout is the most common inflammatory arthritis, affecting ~4% of US adults. It results from deposition of monosodium urate (MSU) crystals in joints and soft tissues when serum urate exceeds the saturation threshold (~6.8 mg/dL).
Uric Acid Metabolism
Uric acid is the end product of purine metabolism (via xanthine oxidase). Approximately two-thirds of urate is excreted renally (via URAT1, GLUT9, ABCG2 transporters) and one-third via the gut. Hyperuricemia results from underexcretion (~90% of cases — renal impairment, thiazides, loop diuretics, low-dose aspirin, cyclosporine) or overproduction (~10% — myeloproliferative disorders, tumor lysis syndrome, Lesch-Nyhan syndrome [HGPRT deficiency], high-purine diet).
Clinical Stages
| Stage | Features | Management |
|---|---|---|
| Asymptomatic hyperuricemia | Elevated serum urate without symptoms | No pharmacologic ULT indicated; lifestyle modification; address cause |
| Acute gout flare | Podagra (1st MTP joint, ~50% of first attacks); dramatic monoarthritis with erythema, warmth, exquisite tenderness; peaks in 12–24 hours | Colchicine, NSAIDs, or corticosteroids (see below) |
| Intercritical period | Asymptomatic between flares; MSU crystals still present | Initiate ULT if recurrent flares (≥2/year), tophi, CKD stage ≥3, urolithiasis |
| Chronic tophaceous gout | Tophi (nodular MSU deposits) in joints, tendons, bursae, ears; chronic arthritis | Aggressive ULT to dissolve tophi; target urate <5 mg/dL |
Acute Flare Management
| Agent | Dose | Notes |
|---|---|---|
| Colchicine | 1.2 mg PO then 0.6 mg 1 hour later (low-dose regimen); within 36 hours of flare onset | Most effective if started within 12–24 hours; dose-adjust for renal impairment; avoid with strong CYP3A4 inhibitors |
| NSAIDs | Indomethacin 50 mg TID or naproxen 500 mg BID (full dose) | Avoid in CKD, GI bleeding, heart failure |
| Corticosteroids | Prednisone 30–40 mg/day x5 days; or intra-articular triamcinolone | Preferred in CKD, polyarticular flares; avoid in poorly controlled diabetes |
| IL-1 inhibitors | Anakinra 100 mg SC daily x3 days (off-label) | Reserved for refractory flares or when NSAIDs/colchicine/steroids contraindicated |
Urate-Lowering Therapy (ULT)
| Drug | Mechanism | Dose | Target | Key Issues |
|---|---|---|---|---|
| Allopurinol | Xanthine oxidase inhibitor | Start 100 mg/day (50 mg if CKD stage ≥3); titrate by 100 mg q2–4 weeks to target | <6 mg/dL (or <5 if tophi) | HLA-B*5801 testing before starting (mandatory in Southeast Asian, African American patients) — risk of severe DRESS/SJS/TEN |
| Febuxostat | Xanthine oxidase inhibitor (non-purine) | 40–80 mg/day | <6 mg/dL | FDA boxed warning: increased CV mortality vs. allopurinol (CARES trial); use if allopurinol contraindicated/intolerant |
| Probenecid | Uricosuric (blocks URAT1) | 500 mg BID, titrate to 2 g/day | <6 mg/dL | Requires adequate renal function (CrCl >50); avoid with urolithiasis history |
| Pegloticase | Recombinant uricase (converts urate to allantoin) | 8 mg IV q2 weeks | Refractory gout | Immunogenic — monitor uric acid before each infusion; discontinue if urate >6 (loss of response, anaphylaxis risk) |
12 Calcium Pyrophosphate Deposition Disease (CPPD)
CPPD (formerly "pseudogout") results from deposition of calcium pyrophosphate dihydrate (CPP) crystals in articular cartilage and synovium. It is predominantly a disease of the elderly — prevalence increases dramatically after age 60.
Clinical Presentations
| Pattern | Features | Key Distinction |
|---|---|---|
| Acute CPP crystal arthritis ("pseudogout") | Acute monoarthritis (knee most common, then wrist); self-limited over 1–3 weeks | Mimics gout but affects different joints; rhomboid crystals |
| Chronic CPP inflammatory arthritis | Chronic polyarthritis resembling RA (MCPs, wrists) | "Pseudo-RA"; look for chondrocalcinosis on X-ray |
| OA with CPPD | Accelerated OA in atypical joints (wrist, MCP, shoulder, ankle) | OA in unusual locations should prompt CPPD evaluation |
| Asymptomatic chondrocalcinosis | Incidental radiographic finding; no treatment needed | Common in elderly — prevalence ~15% at age 65–75, ~40% at >80 |
Diagnosis
Definitive: rhomboid, weakly positively birefringent crystals under compensated polarized light microscopy (blue when parallel to the slow axis of the compensator — opposite to MSU). X-ray: chondrocalcinosis — linear calcification within articular cartilage, most commonly seen in the knee menisci, triangular fibrocartilage of the wrist (TFCC), and pubic symphysis.
Associated Conditions (“The 5 H’s”)
CPPD in a patient <60 years should prompt evaluation for: Hyperparathyroidism, Hemochromatosis, Hypomagnesemia, Hypothyroidism, and Hypophosphatasia. Check calcium, PTH, iron studies/ferritin, magnesium, thyroid function.
Treatment
Acute flare: similar to gout (colchicine, NSAIDs, intra-articular or systemic corticosteroids). Chronic disease: low-dose colchicine 0.6 mg daily or BID; HCQ or MTX for chronic inflammatory CPPD. No disease-modifying therapy exists to dissolve CPP crystals or prevent chondrocalcinosis progression.
13 Hydroxyapatite & Other Crystal Disease
Basic Calcium Phosphate (BCP) / Hydroxyapatite Crystal Disease
Hydroxyapatite crystals are the most common cause of calcific periarthritis/tendinitis, particularly in the supraspinatus tendon (shoulder). These crystals are too small to see on standard polarized light microscopy (require alizarin red staining or electron microscopy). Acute calcific tendinitis presents with severe shoulder pain and X-ray showing calcific deposits in the rotator cuff. Treatment: NSAIDs, local corticosteroid injection, or needle aspiration/lavage of the calcific deposit.
Milwaukee Shoulder Syndrome
A destructive arthropathy of the shoulder (or knee) in elderly women, caused by BCP crystals. Features: large cool effusion (non-inflammatory, WBC <1000), rotator cuff tear, glenohumeral joint destruction. Synovial fluid contains BCP crystals (alizarin red positive) and is often bloody. Essentially a crystal-associated rapidly destructive OA.
Crystal Identification Summary
| Crystal | Shape | Birefringence | Color (Parallel to Compensator) | Disease |
|---|---|---|---|---|
| Monosodium urate (MSU) | Needle-shaped | Strong negative | Yellow when parallel | Gout |
| Calcium pyrophosphate (CPP) | Rhomboid / rod-shaped | Weak positive | Blue when parallel | CPPD (pseudogout) |
| Hydroxyapatite (BCP) | Amorphous clumps | Non-birefringent | Not visible on polarized microscopy | Calcific tendinitis, Milwaukee shoulder |
| Calcium oxalate | Bipyramidal (envelope-shaped) | Positive | Blue when parallel | Dialysis patients, primary hyperoxaluria |
14 Ankylosing Spondylitis
Ankylosing spondylitis (AS) is the prototype axial spondyloarthropathy, characterized by chronic inflammation of the sacroiliac joints and spine leading to progressive ankylosis (bony fusion). It typically begins in males aged 15–30 (M:F = 3:1 for radiographic AS; ratio narrows for non-radiographic axSpA).
HLA-B27 Association
HLA-B27 is present in ~90% of AS patients (vs. ~8% of the general Caucasian population). However, most HLA-B27-positive individuals (~95%) never develop AS. HLA-B27 is neither necessary nor sufficient for diagnosis but supports clinical suspicion. The proposed mechanism involves the "arthritogenic peptide hypothesis" (HLA-B27 presents self-peptides to CD8+ T cells) and endoplasmic reticulum stress from HLA-B27 misfolding, activating IL-23/IL-17 pathways.
Modified New York Criteria (1984)
| Clinical Criteria | Radiographic Criterion |
|---|---|
| 1. Low back pain ≥3 months, improved with exercise, not relieved by rest | Bilateral sacroiliitis grade ≥2 OR unilateral grade ≥3 on X-ray |
| 2. Limitation of lumbar spine motion (sagittal and frontal planes) | |
| 3. Reduced chest expansion (<2.5 cm relative to normal) |
Definite AS = radiographic criterion + ≥1 clinical criterion. Note: These criteria require radiographic changes that may take years to develop, leading to diagnostic delay of 7–10 years. ASAS criteria for axial spondyloarthropathy incorporate MRI (bone marrow edema at SI joints) to enable earlier diagnosis.
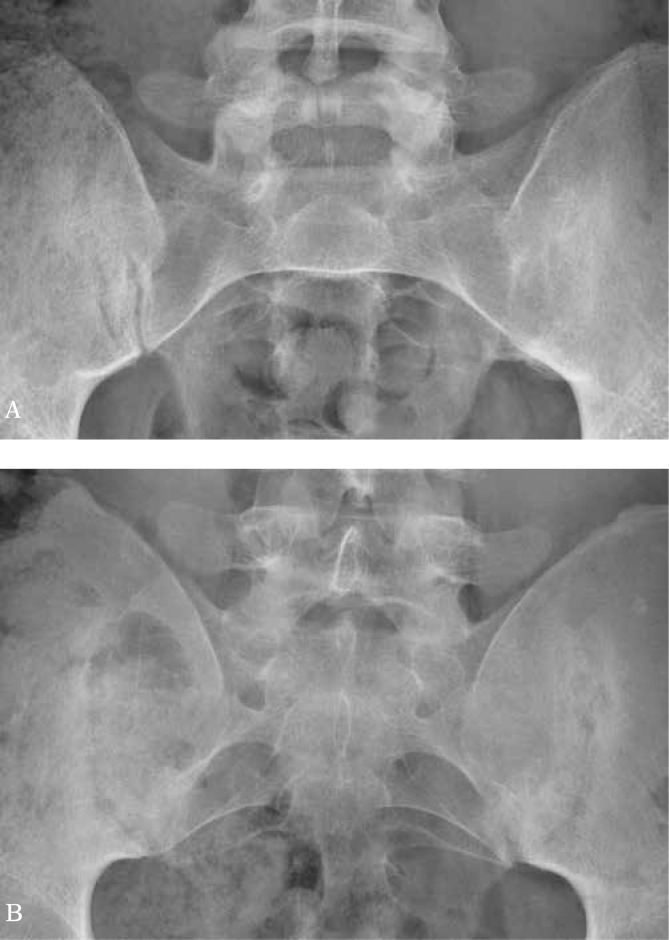
Clinical Features
Inflammatory back pain (onset <40 years, insidious, duration >3 months, improves with exercise, NOT relieved by rest, nocturnal pain with morning stiffness >30 minutes). Progressive spinal fusion: loss of lumbar lordosis → increased thoracic kyphosis → "bamboo spine" (complete fusion of vertebral bodies via syndesmophytes on X-ray). Extra-articular: acute anterior uveitis (the most common extra-articular manifestation, ~25–30%), aortitis/aortic regurgitation, apical pulmonary fibrosis, IgA nephropathy, enthesitis (Achilles, plantar fascia), dactylitis.
Treatment
| Line | Therapy | Details |
|---|---|---|
| First-line | NSAIDs | Continuous full-dose NSAID (e.g., naproxen 500 mg BID, indomethacin 75 mg BID); may slow radiographic progression; try ≥2 NSAIDs before escalating |
| Second-line | TNF inhibitors | Adalimumab 40 mg SC q2 weeks, etanercept 50 mg SC weekly, infliximab 5 mg/kg IV; highly effective for axial and peripheral disease |
| Second-line | IL-17A inhibitors | Secukinumab 150 mg SC monthly, ixekizumab; effective for axial disease; avoid in IBD (can worsen Crohn disease) |
| Third-line | JAK inhibitors | Tofacitinib, upadacitinib (FDA-approved for AS); for inadequate response to biologics |
| Adjunctive | Physical therapy | Essential — spinal extension exercises, swimming; maintain posture and mobility |
15 Psoriatic Arthritis
Psoriatic arthritis (PsA) affects ~30% of psoriasis patients. It can precede skin disease in ~15% of cases. Nail disease is the strongest clinical predictor of PsA development in a psoriasis patient.
CASPAR Classification Criteria
Established inflammatory articular disease (peripheral, axial, or entheseal) PLUS ≥3 points from:
| Feature | Score |
|---|---|
| Current psoriasis | 2 |
| History of psoriasis (if no current psoriasis) | 1 |
| Family history of psoriasis (1st or 2nd degree relative) | 1 |
| Dactylitis (current or history) | 1 |
| Juxta-articular new bone formation (X-ray of hands/feet) | 1 |
| RF negative | 1 |
| Nail dystrophy (pitting, onycholysis, hyperkeratosis) | 1 |
Five Patterns of PsA
| Pattern | Description | Frequency |
|---|---|---|
| Symmetric polyarthritis | RA-like pattern (MCPs, PIPs, wrists) | ~40% |
| Asymmetric oligoarthritis | ≤4 joints; large and small joints | ~30% |
| DIP predominant | DIP joints (associated with nail disease) | ~15% |
| Spondylitis / sacroiliitis | Axial disease, often asymmetric | ~5% |
| Arthritis mutilans | Destructive, osteolytic, "pencil-in-cup" deformity, telescoping digits | <5% |
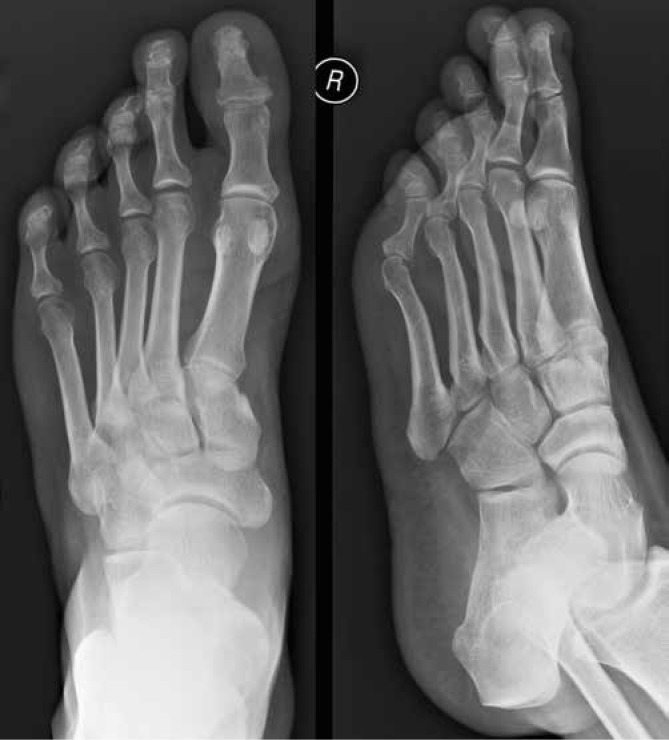
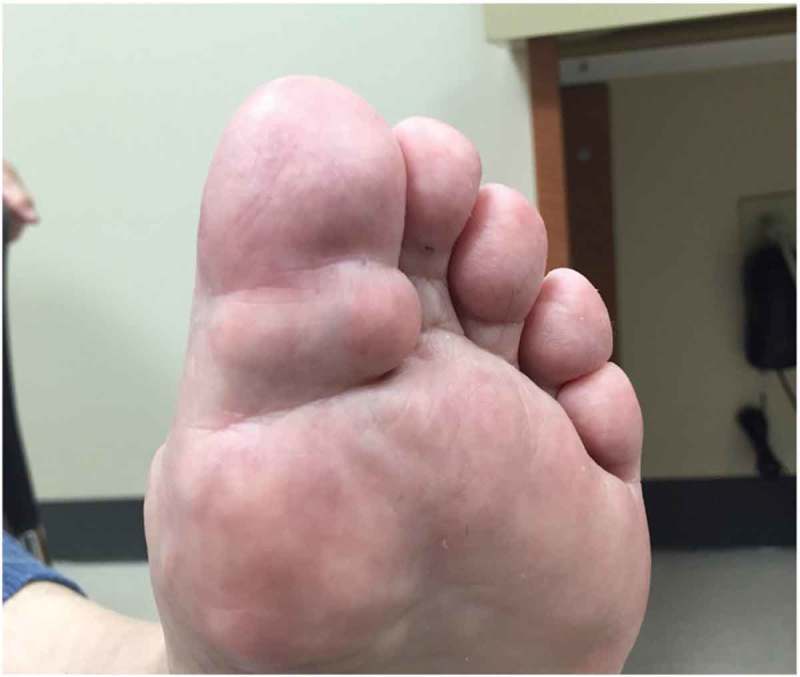
Distinctive Features
Dactylitis ("sausage digit") — diffuse swelling of an entire digit from synovitis + tenosynovitis + enthesitis; highly characteristic. Enthesitis — inflammation at tendon/ligament insertions (Achilles, plantar fascia, patellar tendon). Nail disease — pitting, onycholysis, subungual hyperkeratosis, oil-drop sign; correlates with DIP arthritis (shared entheseal insertion). Radiographic: erosions WITH new bone formation (periostitis, "pencil-in-cup" deformity, ankylosis) — unlike RA which shows erosions only.
Treatment
| Domain | First-Line | If Inadequate Response |
|---|---|---|
| Peripheral arthritis | NSAIDs → MTX, SSZ, or LEF | TNFi, IL-17i (secukinumab, ixekizumab), IL-23i (guselkumab), or PDE4i (apremilast) |
| Axial disease | NSAIDs | TNFi or IL-17i (csDMARDs NOT effective for axial PsA) |
| Enthesitis / dactylitis | NSAIDs | TNFi, IL-17i, or IL-23i |
| Skin | Topical therapy | IL-17i, IL-23i, TNFi (all effective for both skin and joints) |
16 Reactive Arthritis
Reactive arthritis (ReA) is an aseptic inflammatory arthritis triggered by a preceding infection, classically summarized as "can't see, can't pee, can't climb a tree" (conjunctivitis/uveitis, urethritis, arthritis). HLA-B27 is present in 50–80% of cases.
Triggering Infections
| Source | Organisms | Typical Setting |
|---|---|---|
| Genitourinary | Chlamydia trachomatis (most common GU trigger) | Sexually active young adults; may be subclinical |
| Gastrointestinal | Salmonella, Shigella, Yersinia, Campylobacter | Post-diarrheal illness; epidemic or endemic exposure |
Clinical Features
Arthritis: asymmetric oligoarthritis, predominantly lower extremity (knees, ankles, feet), onset 1–4 weeks after infection. Enthesitis: Achilles tendinitis, plantar fasciitis, dactylitis. Urethritis: sterile pyuria. Conjunctivitis (most common ocular manifestation; anterior uveitis in more severe cases). Mucocutaneous: keratoderma blennorrhagicum (psoriasiform lesions on palms/soles, histologically identical to pustular psoriasis), circinate balanitis (painless ulceration on glans penis), oral ulcers.
Management
Most cases are self-limited (3–12 months). NSAIDs are first-line. If Chlamydia-triggered, treat with appropriate antibiotics (azithromycin or doxycycline). For persistent arthritis (>3–6 months): sulfasalazine. For refractory cases: MTX or TNF inhibitors. Intra-articular corticosteroids for persistent monoarthritis. Physical therapy for enthesitis and mobility.
17 Enteropathic Arthritis & IBD-Associated
Joint disease occurs in 10–35% of patients with inflammatory bowel disease (Crohn disease and ulcerative colitis). It is classified into peripheral and axial patterns.
Peripheral Arthritis
| Type | Features | Relationship to Bowel Disease |
|---|---|---|
| Type 1 (pauciarticular) | <5 large joints (knees, ankles); acute, migratory; self-limited (<10 weeks) | Parallels IBD activity — flares with bowel flares; improves with IBD treatment |
| Type 2 (polyarticular) | ≥5 small joints (MCPs, PIPs); chronic, symmetric | Independent of IBD activity — runs its own course |
Axial Disease
Sacroiliitis and spondylitis occur in 5–10% of IBD patients. Unlike AS, IBD-associated sacroiliitis is often asymmetric. Axial disease runs independently of bowel disease activity. HLA-B27 is positive in ~50% (vs. 90% in primary AS).
Treatment Considerations
NSAIDs should be used cautiously (may exacerbate IBD). Sulfasalazine treats both peripheral arthritis and mild colitis. TNF inhibitors (infliximab, adalimumab) are effective for both IBD and arthritis — avoid etanercept (less effective for IBD). IL-17 inhibitors are contraindicated in IBD (can trigger or worsen Crohn disease). Anti-IL-12/23 (ustekinumab) is effective for both Crohn disease and SpA.
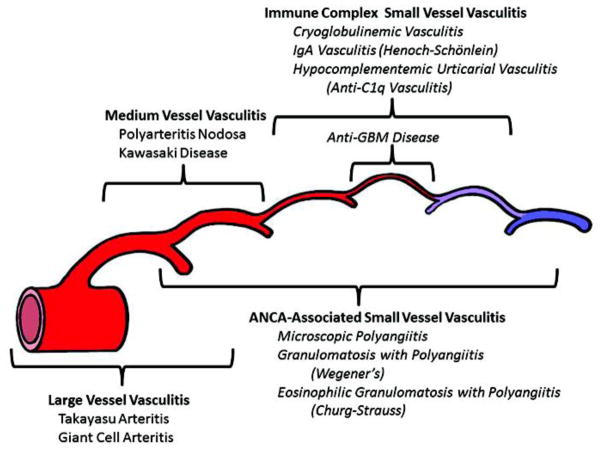
18 Large Vessel Vasculitis
Giant Cell Arteritis (GCA)
GCA is the most common systemic vasculitis in adults >50 years. It affects the aorta and its branches, with a predilection for the extracranial branches of the carotid artery (superficial temporal artery). Nearly always occurs after age 50; average onset ~72 years. Closely associated with polymyalgia rheumatica (PMR) — 40–60% of GCA patients have PMR; 15–20% of PMR patients develop GCA.
GCA is a medical emergency when vision is threatened. Anterior ischemic optic neuropathy (AION) causes sudden, painless, irreversible monocular vision loss. Without treatment, the contralateral eye is at high risk (>50% within days to weeks). Start high-dose corticosteroids IMMEDIATELY upon clinical suspicion — do NOT wait for biopsy results. Dose: methylprednisolone 1000 mg IV daily x3 days if visual symptoms present, then prednisone 1 mg/kg/day (max 60–80 mg).
Diagnosis
| Method | Details |
|---|---|
| Temporal artery biopsy | Gold standard; obtain ≥1 cm specimen (skip lesions can cause false negatives); biopsy within 2 weeks of starting steroids (histology remains positive); contralateral biopsy if first negative increases yield by 5–10% |
| Temporal artery ultrasound | "Halo sign" (hypoechoic circumferential thickening); increasingly used as first-line in experienced centers |
| Laboratory | ESR markedly elevated (often >50–100); CRP elevated; normocytic anemia; thrombocytosis |
Treatment
High-dose prednisone (40–60 mg/day without visual symptoms; IV pulse then 1 mg/kg if visual symptoms) with slow taper over 12–24 months. Tocilizumab (IL-6 receptor inhibitor) is FDA-approved as a steroid-sparing agent for GCA (GiACTA trial: sustained remission at 52 weeks: 56% with tocilizumab vs. 14% with prednisone alone). Low-dose aspirin may reduce ischemic complications.
Takayasu Arteritis
Takayasu arteritis affects the aorta and its major branches, predominantly in women <40 years (F:M = 9:1). "Pulseless disease" — inflammation causes stenosis, occlusion, and aneurysm formation. Clinical: limb claudication, blood pressure discrepancy between arms (>10 mmHg), absent/diminished pulses, bruits, constitutional symptoms. Diagnosis: CTA or MRA showing vessel wall thickening, stenoses, and aneurysms. Treatment: corticosteroids (1 mg/kg/day) ± MTX, AZA, or tocilizumab as steroid-sparing agents. Surgical bypass or angioplasty for critical stenoses in quiescent disease.
19 Medium Vessel Vasculitis
Polyarteritis Nodosa (PAN)
PAN is a necrotizing vasculitis of medium-sized muscular arteries, typically sparing small vessels and glomeruli (distinguishes from ANCA-associated vasculitis). Associated with hepatitis B in ~10–30% of cases. ANCA is NEGATIVE in PAN.
| System | Manifestation | Mechanism |
|---|---|---|
| Constitutional | Fever, weight loss, malaise | Systemic inflammation |
| Renal | Renovascular hypertension, renal infarction | Renal artery aneurysms and stenoses; NOT glomerulonephritis |
| GI | Abdominal pain, GI bleeding, bowel infarction | Mesenteric artery vasculitis |
| Neurologic | Mononeuritis multiplex (most common), CNS rarely | Vasa nervorum involvement |
| Skin | Livedo reticularis, skin nodules, ulcers, digital gangrene | Cutaneous arterial inflammation |
| Musculoskeletal | Myalgias, arthralgias | Muscle arterial involvement |
| Testicular | Orchitis (testicular pain) | Testicular artery vasculitis |
Diagnosis: Angiography showing microaneurysms and segmental stenoses ("string of beads") in renal, mesenteric, or hepatic arteries. Biopsy (sural nerve, affected skin) shows fibrinoid necrosis of medium artery walls. Treatment: corticosteroids + cyclophosphamide for severe disease; treat underlying hepatitis B if present (antiviral therapy + plasma exchange).
Kawasaki Disease
Kawasaki disease is an acute, self-limited vasculitis of childhood (peak age 6 months to 5 years), important because of coronary artery aneurysm risk (~25% if untreated, <5% with treatment). Diagnosis: fever ≥5 days PLUS ≥4 of 5 criteria: (1) bilateral conjunctival injection (non-exudative), (2) oral changes (strawberry tongue, lip fissures), (3) cervical lymphadenopathy (≥1.5 cm, usually unilateral), (4) polymorphous rash, (5) extremity changes (edema, erythema, desquamation). Treatment: IVIG 2 g/kg single infusion + high-dose aspirin (anti-inflammatory, then low-dose anti-platelet after defervescence). Echocardiographic monitoring for coronary aneurysms.
20 ANCA-Associated Vasculitis
The ANCA-associated vasculitides (AAV) are small-vessel necrotizing vasculitides characterized by pauci-immune (little or no immunoglobulin deposition) glomerulonephritis and systemic inflammation. The three entities are GPA, MPA, and EGPA.
ANCA Patterns & Targets
| ANCA Pattern | Target Antigen | Disease Association |
|---|---|---|
| c-ANCA (cytoplasmic) | Proteinase 3 (PR3) | GPA (~90%) |
| p-ANCA (perinuclear) | Myeloperoxidase (MPO) | MPA (~60–70%), EGPA (~40%), drug-induced vasculitis |
Disease Characteristics
| Feature | GPA | MPA | EGPA |
|---|---|---|---|
| Upper airway | Yes (sinusitis, nasal crusting, saddle-nose deformity, subglottic stenosis) | No | Allergic rhinitis, nasal polyps |
| Lower airway | Pulmonary nodules, cavitary lesions, alveolar hemorrhage | Alveolar hemorrhage | Asthma (severe, late-onset), eosinophilic infiltrates |
| Renal | Pauci-immune crescentic GN (RPGN) | Pauci-immune crescentic GN (most common cause of pulmonary-renal syndrome) | GN less common (~25%) |
| Neurologic | Cranial nerve palsies | Mononeuritis multiplex | Mononeuritis multiplex (most common in EGPA) |
| Eosinophilia | No | No | Yes (peripheral eosinophilia >10%, tissue eosinophilia) |
| Granulomas | Yes | No | Yes (eosinophilic) |
| ANCA | c-ANCA/PR3 (~90%) | p-ANCA/MPO (~60–70%) | p-ANCA/MPO (~40%); ANCA-negative in ~60% |
Diffuse alveolar hemorrhage (DAH) + rapidly progressive glomerulonephritis (RPGN) is a life-threatening emergency. Patients present with hemoptysis (may be absent initially), dropping hemoglobin, bilateral infiltrates, and acute kidney injury with active urine sediment (RBC casts, dysmorphic RBCs). Immediate treatment: pulse IV methylprednisolone 500–1000 mg x3 days + rituximab (375 mg/m² weekly x4 or 1000 mg x2) or cyclophosphamide. Consider plasma exchange for severe renal disease (creatinine >5.7 mg/dL) or DAH with respiratory failure. Check ANCA, anti-GBM (rule out Goodpasture syndrome).
Treatment of AAV
| Phase | Regimen | Duration |
|---|---|---|
| Induction (severe/organ-threatening) | Rituximab 375 mg/m² IV weekly x4 (preferred per RAVE/RITUXVAS) OR CYC (IV pulse: 15 mg/kg q2–4 weeks x3–6 months, or oral 2 mg/kg/day) + glucocorticoids + avacopan (C5a receptor inhibitor, steroid-sparing) | 3–6 months |
| Induction (limited/non-organ-threatening) | MTX 25 mg/week + glucocorticoids (for GPA without renal involvement) | 3–6 months |
| Maintenance | Rituximab 500 mg IV q6 months (preferred per MAINRITSAN) OR AZA 2 mg/kg/day OR MMF | ≥24 months (longer for PR3-ANCA positive, relapsing disease) |
21 Small Vessel Vasculitis
IgA Vasculitis (Henoch-Schönlein Purpura)
IgA vasculitis (IgAV) is the most common vasculitis in children (peak age 3–10 years). It is mediated by IgA immune complex deposition in small vessels. Classic tetrad: (1) palpable purpura (non-thrombocytopenic, gravitationally dependent — lower extremities and buttocks), (2) arthritis/arthralgias (knees, ankles; non-deforming), (3) GI involvement (abdominal pain, GI bleeding, intussusception risk in children), (4) renal disease (IgA nephropathy — hematuria, proteinuria; identical histology to primary IgA nephropathy). Usually self-limited; treat with supportive care. Corticosteroids for severe GI or renal involvement.
Cryoglobulinemic Vasculitis
Cryoglobulins are immunoglobulins that precipitate at cold temperatures and re-dissolve with warming.
| Type | Composition | Association |
|---|---|---|
| Type I | Monoclonal IgM or IgG | Lymphoproliferative disorders (Waldenström, myeloma) |
| Type II (mixed) | Monoclonal IgM (with RF activity) + polyclonal IgG | Hepatitis C (most common cause of mixed cryoglobulinemia) |
| Type III (mixed) | Polyclonal IgM + polyclonal IgG | Autoimmune diseases, infections |
Clinical triad of mixed cryoglobulinemia: purpura, arthralgias, weakness (Meltzer triad). Also: membranoproliferative GN, peripheral neuropathy. Low C4 (classic pathway consumption) with normal C3 is characteristic. Treatment: treat underlying hepatitis C (direct-acting antivirals); rituximab for severe vasculitis; plasma exchange for life-threatening disease.
Hypersensitivity Vasculitis (Cutaneous Leukocytoclastic Vasculitis)
Isolated small vessel vasculitis limited to skin. Triggered by drugs (antibiotics, NSAIDs), infections, or idiopathic. Presents with palpable purpura on lower extremities. Skin biopsy shows leukocytoclastic vasculitis (fibrinoid necrosis, neutrophilic infiltrate, nuclear dust). Usually self-limited; remove offending agent. Rule out systemic vasculitis with urinalysis, creatinine, complement levels.
22 Behçet Disease
Behçet disease is a systemic vasculitis of variable vessel size, most prevalent along the ancient Silk Road (Turkey, Iran, Japan, China). It is the only vasculitis that can affect both arteries and veins of all sizes. HLA-B51 is associated (~60% of patients in endemic areas).
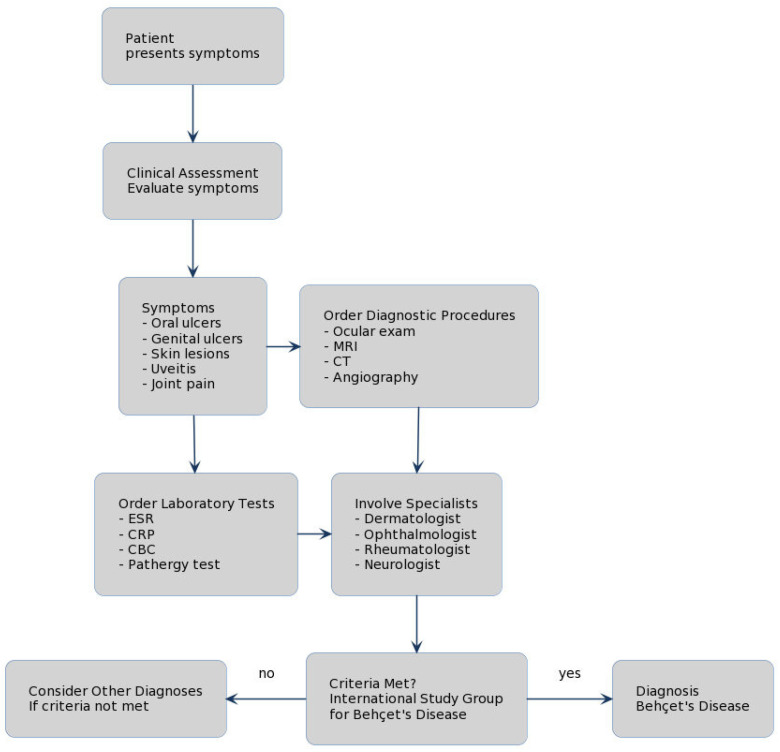
Clinical Features
| Manifestation | Features | Frequency |
|---|---|---|
| Oral ulcers | Recurrent (≥3 times/year), painful aphthous ulcers; the most common and often earliest manifestation | >95% |
| Genital ulcers | Painful ulcers on scrotum/vulva; often leave scars (distinguishing from oral aphthae) | 60–90% |
| Ocular | Anterior/posterior uveitis, retinal vasculitis; can cause blindness | 50–70% |
| Skin | Erythema nodosum, pseudofolliculitis, papulopustular lesions, pathergy | 40–80% |
| Vascular | DVT, superficial thrombophlebitis, arterial aneurysms (pulmonary artery aneurysm is characteristic) | 25–40% |
| Neurologic (neuro-Behçet) | Brainstem syndrome, meningoencephalitis, cerebral venous sinus thrombosis | 5–10% |
| Arthritis | Non-erosive oligoarthritis (knees, ankles) | 40–60% |
Pathergy Test
A skin prick with a sterile needle producing a papule or pustule ≥2 mm at 24–48 hours. Positive pathergy is relatively specific for Behçet disease but sensitivity varies geographically (positive in ~60% of Turkish patients, rare in Western populations).
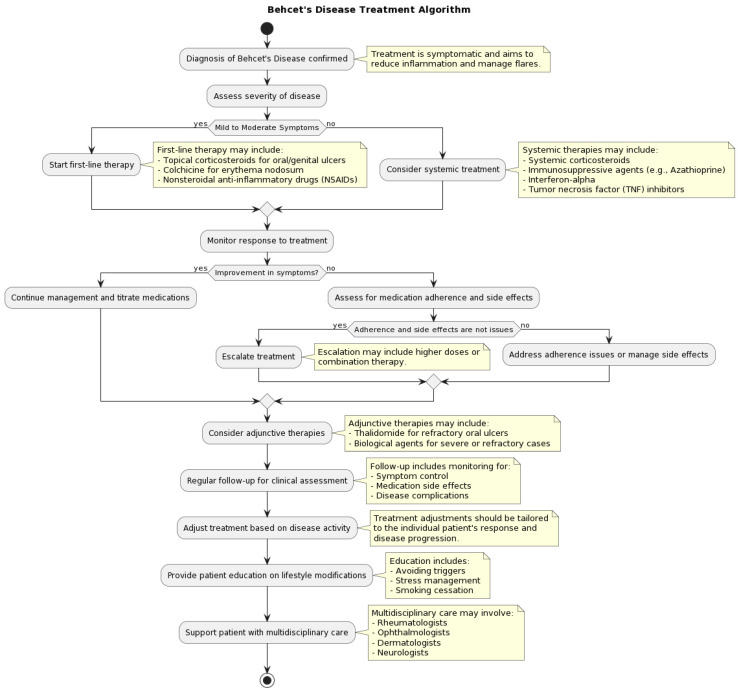
Treatment
Oral ulcers: topical corticosteroids, colchicine 0.5–1 mg BID. Ocular disease: systemic corticosteroids + AZA or interferon-alpha; infliximab or adalimumab for sight-threatening uveitis (rapid response). Vascular: anticoagulation is controversial for thrombosis (risk of aneurysm rupture); immunosuppression (AZA, CYC) is more important. Neuro-Behçet: corticosteroids + AZA or CYC; TNFi for refractory. Apremilast is FDA-approved for oral ulcers in Behçet.
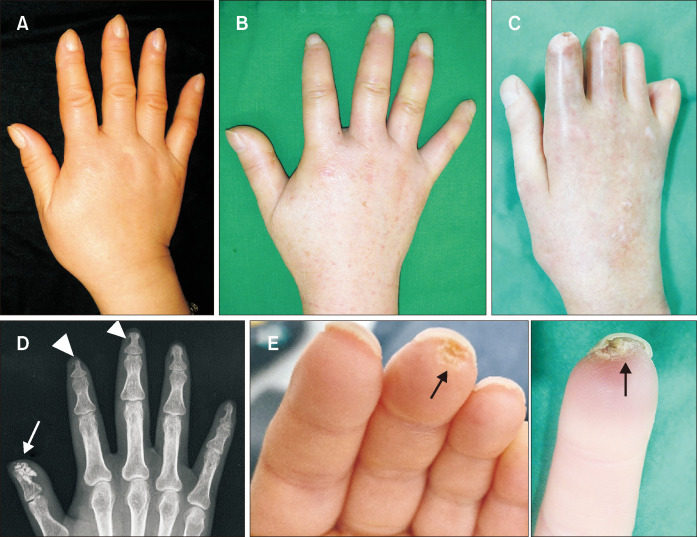
23 Systemic Sclerosis
Systemic sclerosis (SSc/scleroderma) is characterized by vascular injury, immune activation, and progressive fibrosis of the skin and internal organs. F:M = 4:1; peak onset 30–50 years. It has the highest case-specific mortality of all rheumatic diseases.
Limited vs. Diffuse SSc
| Feature | Limited Cutaneous SSc | Diffuse Cutaneous SSc |
|---|---|---|
| Skin involvement | Distal to elbows/knees + face | Proximal to elbows/knees + trunk |
| Raynaud onset to skin | Years to decades | Within 1 year |
| Antibody | Anticentromere | Anti-Scl-70 (anti-topoisomerase I) |
| Organ complications | Pulmonary arterial hypertension (PAH), primary biliary cholangitis | Interstitial lung disease (ILD), scleroderma renal crisis, cardiac disease |
| CREST syndrome | Calcinosis, Raynaud, Esophageal dysmotility, Sclerodactyly, Telangiectasia | Not typical |
| Prognosis | Better; 10-year survival ~75% | Worse; 10-year survival ~55% |
Key Antibodies
| Antibody | SSc Subtype | Associated Complication |
|---|---|---|
| Anticentromere | Limited | PAH, limited skin disease |
| Anti-Scl-70 (topoisomerase I) | Diffuse | ILD, diffuse skin disease |
| Anti-RNA polymerase III | Diffuse | Scleroderma renal crisis; malignancy association |
| Anti-U3 RNP (fibrillarin) | Diffuse | PAH, skeletal muscle involvement |
| Anti-PM-Scl | Overlap | Myositis-scleroderma overlap |
Occurs in 10–15% of diffuse SSc, particularly within first 4 years and in those receiving high-dose corticosteroids (>15 mg/day prednisone). Presents with severe hypertension (accelerated/malignant), acute kidney injury (rising creatinine over days), microangiopathic hemolytic anemia (schistocytes, elevated LDH, low haptoglobin), and thrombocytopenia. Treatment: ACE inhibitors (captopril) — titrate to normalize BP; do NOT withhold ACEi even if creatinine rises initially. ACEi have transformed prognosis from ~10% to ~60% 1-year survival. Avoid corticosteroids >15 mg/day in diffuse SSc — major risk factor for renal crisis.
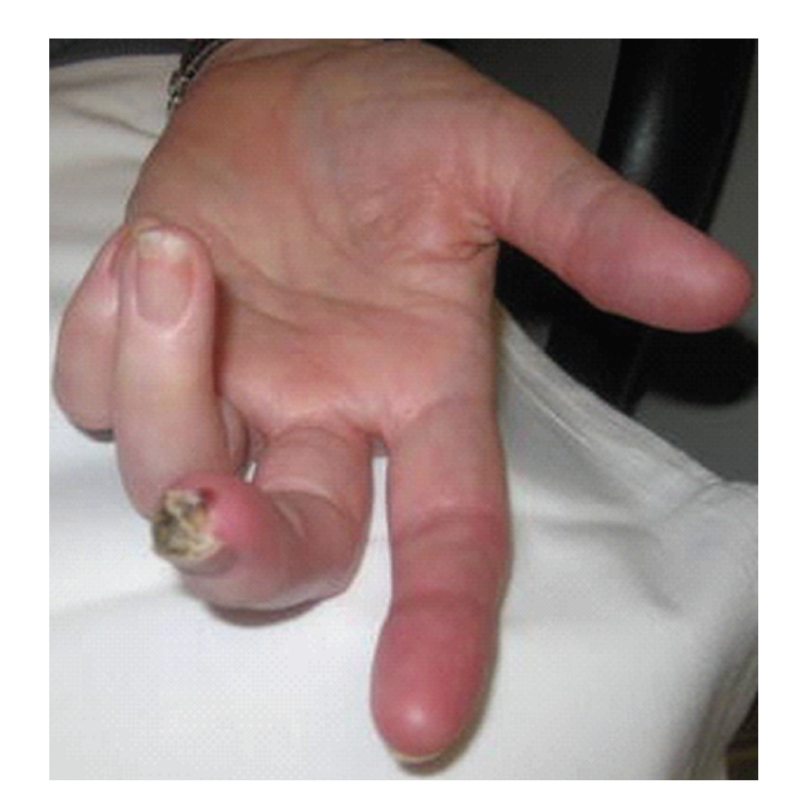
Screening & Monitoring
ILD screening: baseline and annual PFTs (FVC, DLCO) + HRCT at diagnosis. Anti-Scl-70 positive and male sex are risk factors. Treat with mycophenolate or nintedanib for progressive ILD. PAH screening: annual echocardiography with DLCO. Anticentromere antibody and isolated DLCO decline are risk factors. Right heart catheterization to confirm. Treat with PDE5 inhibitors, endothelin receptor antagonists (bosentan, ambrisentan), prostacyclin analogs, riociguat.
24 Inflammatory Myopathies
The idiopathic inflammatory myopathies (IIM) are a heterogeneous group of autoimmune diseases targeting skeletal muscle. The major subtypes are dermatomyositis (DM), polymyositis (PM), inclusion body myositis (IBM), anti-synthetase syndrome, and immune-mediated necrotizing myopathy (IMNM).
Clinical Comparison
| Feature | Dermatomyositis | Polymyositis | Inclusion Body Myositis |
|---|---|---|---|
| Demographics | Adults or children; F > M | Adults >18 years; F > M | Men >50 years (most common IIM in >50) |
| Weakness pattern | Symmetric proximal | Symmetric proximal | Asymmetric; proximal AND distal (finger flexors, quadriceps) |
| Skin findings | Gottron papules, heliotrope rash, V-sign, shawl sign, mechanic's hands | None | None |
| CK level | Elevated (10–50×) | Very elevated (10–100×) | Mildly elevated (1–10×) or normal |
| Malignancy risk | High (15–25%) — screen at diagnosis | Moderate (10–15%) | Not increased |
| Response to immunosuppression | Good | Good | Poor (no effective treatment) |
| Pathology | Perifascicular atrophy, perivascular inflammation | Endomysial CD8+ T-cell infiltrate | Rimmed vacuoles, endomysial inflammation, amyloid deposits |
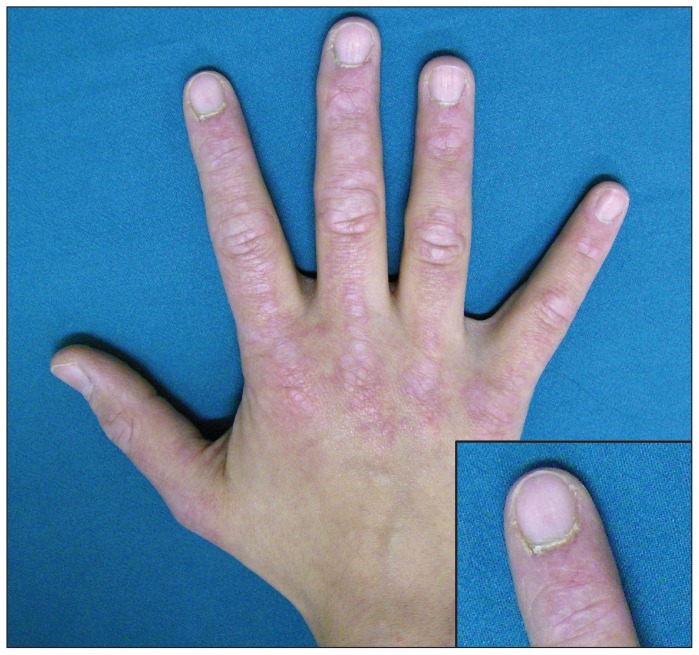
Myositis-Specific Antibodies
| Antibody | Disease | Clinical Association |
|---|---|---|
| Anti-Jo-1 (and other anti-aminoacyl-tRNA synthetases) | Anti-synthetase syndrome | ILD, Raynaud, mechanic's hands, fever, non-erosive arthritis |
| Anti-Mi-2 | Dermatomyositis | Classic DM skin findings; good prognosis, good treatment response |
| Anti-MDA5 (CADM-140) | Clinically amylopathic DM | Rapidly progressive ILD (high mortality); skin findings without significant myopathy |
| Anti-TIF1-gamma (p155/140) | Dermatomyositis | Strongest malignancy association in adult DM |
| Anti-NXP-2 (MJ) | Dermatomyositis | Calcinosis (juvenile DM); malignancy (adult DM) |
| Anti-SRP | Immune-mediated necrotizing myopathy | Severe weakness, very high CK, cardiac involvement; poor response to therapy |
| Anti-HMGCR | Immune-mediated necrotizing myopathy | Statin-associated (but persists after statin discontinuation); requires immunosuppression |
| Anti-cN1A (anti-NT5C1A) | Inclusion body myositis | Sensitive and specific for IBM |
Treatment
DM/PM: high-dose corticosteroids (prednisone 1 mg/kg/day) + steroid-sparing agent (MTX or AZA preferred). For refractory: IVIG (especially effective in DM), rituximab, MMF, or tacrolimus. For anti-MDA5-associated rapidly progressive ILD: aggressive combination immunosuppression (steroids + calcineurin inhibitor + CYC). IBM: no proven effective treatment; IVIG may provide modest temporary benefit; physical therapy is essential.
25 Sjögren Syndrome
Sjögren syndrome (SS) is a chronic autoimmune disease characterized by lymphocytic infiltration of exocrine glands, causing sicca symptoms (dry eyes/dry mouth). It is the second most common autoimmune rheumatic disease after RA. F:M = 9:1; peak onset 40–60 years.
Classification
Primary SS: occurs alone. Secondary SS: occurs in association with another autoimmune disease (RA, SLE, SSc).
Clinical Features
| Domain | Manifestation | Details |
|---|---|---|
| Ocular | Keratoconjunctivitis sicca (dry eyes) | Gritty/sandy sensation, burning, photosensitivity; corneal damage if untreated |
| Oral | Xerostomia (dry mouth) | Difficulty swallowing dry food, dental caries, oral candidiasis, parotid gland enlargement |
| Systemic | Fatigue (most common systemic symptom) | Often debilitating; major driver of disability |
| Articular | Non-erosive arthritis/arthralgias | Small joints; can mimic early RA |
| Pulmonary | Airway dryness, ILD, lymphocytic interstitial pneumonia (LIP) | Dry cough; LIP is characteristic of SS |
| Renal | Interstitial nephritis, distal RTA (type 1) | Hypokalemia, non-anion gap metabolic acidosis |
| Neurologic | Peripheral neuropathy (sensory > motor), CNS disease (rare) | Small fiber neuropathy causes painful burning |
| Hematologic | Lymphoma (4–7% lifetime risk) | B-cell NHL (MALT lymphoma most common); risk factors: persistent parotid enlargement, lymphadenopathy, declining complement, rising RF, purpura, cryoglobulinemia |
Diagnosis
| Test | Purpose | Details |
|---|---|---|
| Anti-SSA/Ro | Most sensitive antibody (60–70%) | Also positive in SLE, neonatal lupus, SCLE |
| Anti-SSB/La | More specific (30–40%) | Rarely positive without SSA; positive SSA + SSB strongly suggests primary SS |
| Schirmer test | Tear production | <5 mm wetting in 5 minutes = abnormal (filter paper in lower conjunctival sac) |
| Ocular staining score | Corneal/conjunctival damage | Lissamine green or fluorescein staining |
| Salivary gland biopsy (minor) | Histopathology | Focus score ≥1 (aggregate of ≥50 lymphocytes per 4 mm²); most specific test |
| Salivary flow rate | Unstimulated whole saliva | ≤0.1 mL/min = abnormal |
26 Mixed Connective Tissue Disease & Overlap Syndromes
Mixed connective tissue disease (MCTD) features overlapping findings of SLE, SSc, and PM with high-titer anti-U1 RNP antibody. It was originally described as a benign overlap syndrome, but it can evolve into a dominant phenotype (most commonly scleroderma) and can cause serious organ damage, especially pulmonary arterial hypertension.
Clinical Features
| Feature | Frequency | Notes |
|---|---|---|
| Raynaud phenomenon | >90% | Often the earliest manifestation |
| Swollen "puffy" hands | ~85% | Characteristic early finding |
| Arthritis | ~80% | Non-erosive, can be deforming (Jaccoud-type) |
| Myositis | ~50–70% | Elevated CK, proximal weakness |
| Esophageal dysmotility | ~50% | Similar to SSc |
| Pulmonary arterial hypertension | 10–25% | Leading cause of death in MCTD; screen with annual echo + DLCO |
| Sclerodactyly | ~40% | Skin thickening limited to digits |
| Serositis | ~30% | Pericarditis, pleuritis (SLE-like) |
Diagnosis
High-titer anti-U1 RNP antibody (essential — must be present) + clinical features of at least 2 of 3 diseases (SLE, SSc, PM). ANA is positive (speckled pattern). Anti-dsDNA and anti-Sm are negative (their presence shifts the diagnosis toward SLE).
Undifferentiated Connective Tissue Disease (UCTD)
Patients with ANA positivity and features suggestive of a CTD but not meeting criteria for any specific disease. Approximately 30% will evolve into a defined CTD (most commonly SLE or SS) over 3–5 years. HCQ is often used empirically. Close follow-up with serial serologies and clinical reassessment is essential.
27 Osteoarthritis
Osteoarthritis (OA) is the most common joint disease worldwide, affecting >300 million people globally. It is a disease of the entire joint — cartilage loss, subchondral bone remodeling, osteophyte formation, synovial inflammation, and ligament/meniscal degeneration.
Mechanical vs. Inflammatory Arthritis
| Feature | OA (Mechanical) | Inflammatory (RA, PsA) |
|---|---|---|
| Morning stiffness | <30 minutes ("gelling") | >60 minutes |
| Pain pattern | Worsens with activity, improves with rest | Improves with activity; worse after rest |
| Joint involvement | DIPs, PIPs, 1st CMC, hips, knees, spine | MCPs, PIPs, wrists, MTPs (RA); DIPs (PsA) |
| Swelling type | Bony enlargement (hard) | Boggy synovial swelling (soft) |
| Systemic symptoms | Absent | Fatigue, malaise, fever |
| Laboratory | Normal ESR/CRP, negative RF/ANA | Elevated ESR/CRP, positive serologies |
| Synovial fluid | Non-inflammatory (<2000 WBC/mm³) | Inflammatory (2000–50,000 WBC/mm³) |
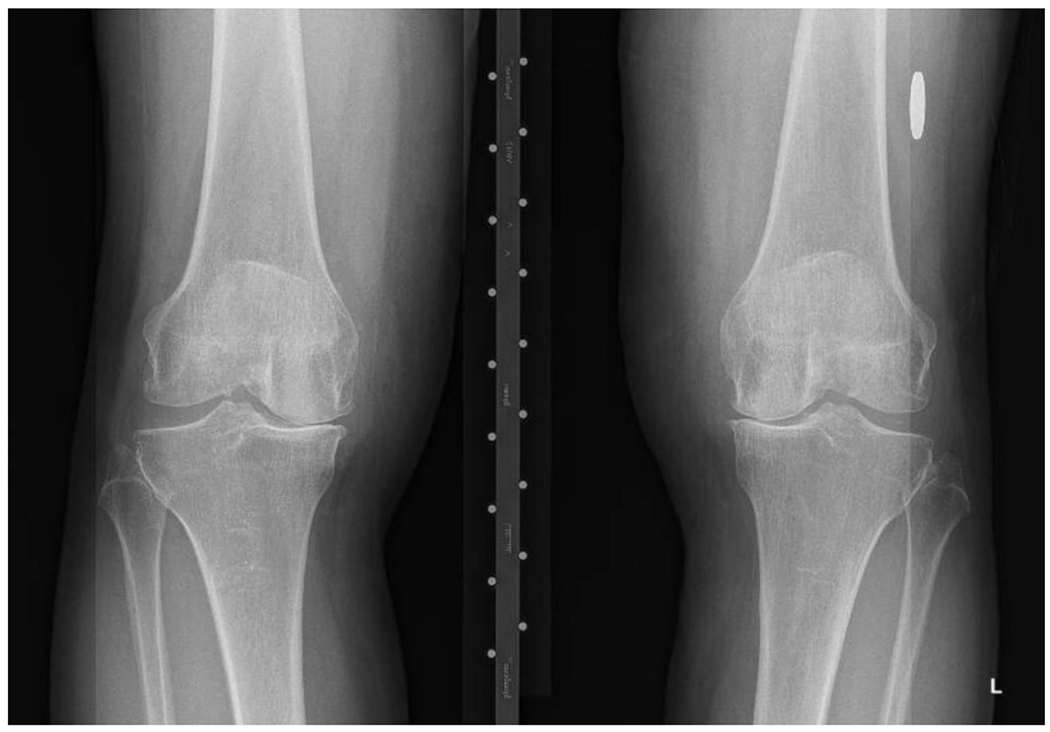
Characteristic Findings
Heberden nodes (bony enlargement at DIP joints) and Bouchard nodes (at PIP joints). First CMC (thumb base) OA causes "squaring" of the hand base. Radiographic features: asymmetric joint space narrowing, subchondral sclerosis, osteophytes, subchondral cysts. Erosive OA (inflammatory variant of hand OA) causes central erosions at DIP/PIP joints with a "gull-wing" or "sawtooth" pattern on X-ray.
Management Ladder
| Step | Intervention |
|---|---|
| Core (all patients) | Weight loss (5–10% body weight reduces knee pain significantly), exercise (low-impact aerobic + strengthening), physical therapy, patient education |
| Pharmacologic first-line | Topical NSAIDs (diclofenac gel — preferred in patients >75), oral acetaminophen (modest benefit), oral NSAIDs (lowest dose, shortest duration) |
| Intra-articular | Corticosteroid injection (short-term relief, 4–6 weeks; limit to 3–4/year per joint); hyaluronic acid injection (modest benefit, controversial) |
| Adjunctive | Duloxetine (centralized pain component), bracing/orthotics, assistive devices |
| Surgical | Total joint arthroplasty (TJA) for refractory knee or hip OA with functional limitation |
28 Fibromyalgia
Fibromyalgia (FM) is a centralized pain syndrome characterized by widespread musculoskeletal pain, fatigue, sleep disturbance, and cognitive dysfunction. It affects 2–8% of the population (F:M = 2:1). It is NOT an inflammatory or autoimmune disease — it is a disorder of central pain processing (central sensitization).
2016 ACR Revised Diagnostic Criteria
| Criterion | Details |
|---|---|
| Widespread Pain Index (WPI) | Count of 19 body areas with pain in the past week (score 0–19) |
| Symptom Severity Score (SSS) | Fatigue, waking unrefreshed, cognitive symptoms (each 0–3) + somatic symptoms score (0–3); total 0–12 |
| Diagnostic threshold | WPI ≥7 AND SSS ≥5 OR WPI 4–6 AND SSS ≥9 |
| Duration | Symptoms present for ≥3 months |
| Exclusion | No other diagnosis that would sufficiently explain the pain |
Pharmacologic Treatment
| Drug Class | Agents | Mechanism | Notes |
|---|---|---|---|
| SNRIs | Duloxetine 60 mg/day; milnacipran 50 mg BID | Serotonin and norepinephrine reuptake inhibition (descending pain inhibition) | FDA-approved for FM; also helps comorbid depression |
| Alpha-2-delta ligands | Pregabalin 150–225 mg BID | Calcium channel modulation; reduces neuronal excitability | FDA-approved for FM; also helps sleep; weight gain, dizziness |
| Tricyclic antidepressants | Amitriptyline 10–50 mg at bedtime | Multiple (serotonin/NE reuptake, sodium channel blockade) | Not FDA-approved but widely used; improves pain and sleep |
| Cyclobenzaprine | 5–10 mg at bedtime | Muscle relaxant (structurally related to TCAs) | Improves sleep and pain; use low-dose |
Non-Pharmacologic Treatment
Exercise is the most effective intervention (aerobic exercise 30 minutes, 3–5 times/week; evidence level A). Cognitive behavioral therapy (CBT) addresses maladaptive pain coping. Sleep hygiene is essential. Patient education about the nature of centralized pain.
29 Adult-Onset Still Disease
Adult-onset Still disease (AOSD) is a systemic inflammatory disorder characterized by a triad of quotidian (daily) spiking fevers, evanescent salmon-pink macular rash, and arthritis. It is considered a diagnosis of exclusion. Bimodal age distribution (15–25 and 36–46 years).
Yamaguchi Criteria (Most Sensitive)
Requires ≥5 criteria (including ≥2 major) after excluding infection, malignancy, and other autoimmune diseases:
| Major Criteria | Minor Criteria |
|---|---|
| Fever ≥39°C, lasting ≥1 week, quotidian pattern | Sore throat |
| Arthralgia or arthritis, ≥2 weeks | Lymphadenopathy |
| Typical rash (salmon-colored, evanescent, macular) | Hepatomegaly or splenomegaly |
| WBC ≥10,000/μL with ≥80% granulocytes | Abnormal LFTs (not attributable to drugs/other) |
| Negative ANA and RF |
Key Laboratory Findings
Ferritin is markedly elevated (often >1000 ng/mL, sometimes >10,000). A glycosylated ferritin fraction ≤20% is relatively specific for AOSD (normal is ~50–80%; low glycosylation suggests ferritin is from inflammatory macrophage production rather than hepatic). High WBC (neutrophilia), elevated ESR/CRP, elevated LFTs, high D-dimer. ANA and RF are characteristically NEGATIVE.
Treatment
| Line | Therapy | Notes |
|---|---|---|
| First-line | NSAIDs + corticosteroids (prednisone 0.5–1 mg/kg/day) | NSAIDs alone effective in mild disease; steroids for systemic features |
| Second-line | IL-1 inhibitors (anakinra 100 mg SC daily; canakinumab 4 mg/kg SC q4 weeks) | Highly effective; rapid response; increasingly used as first-line steroid-sparing |
| Alternatives | IL-6 inhibitor (tocilizumab); MTX; csDMARDs | Tocilizumab for steroid-dependent disease; MTX for arthritis-predominant |
MAS is a life-threatening complication of AOSD (and systemic JIA) — a form of secondary hemophagocytic lymphohistiocytosis (HLH). Features: high fever, hepatosplenomegaly, pancytopenia, markedly elevated ferritin (>10,000), high triglycerides, low fibrinogen, elevated soluble IL-2 receptor (sCD25). Paradoxical fall in ESR (due to fibrinogen consumption) with rising ferritin is a classic clue. Treatment: high-dose corticosteroids, cyclosporine, anakinra; etoposide-based HLH protocols for refractory cases.
30 Joint Aspiration & Injection
Arthrocentesis (joint aspiration) is one of the most important diagnostic AND therapeutic procedures in rheumatology. Synovial fluid analysis can definitively diagnose crystal arthritis and septic arthritis.
Indications
Diagnostic: any unexplained monoarthritis (especially acute), suspected septic arthritis, suspected crystal arthritis, differentiation of inflammatory vs. non-inflammatory effusion. Therapeutic: large tense effusion, intra-articular corticosteroid injection. The only absolute contraindication is overlying cellulitis/soft tissue infection at the needle entry site.
Synovial Fluid Analysis
| Parameter | Normal | Non-Inflammatory (OA) | Inflammatory | Septic |
|---|---|---|---|---|
| Appearance | Clear, colorless | Clear, yellow | Cloudy, yellow | Purulent, opaque |
| Viscosity | High | High | Low | Very low |
| WBC (/mm³) | <200 | <2,000 | 2,000–50,000 | >50,000 (often >100,000) |
| PMN % | <25% | <25% | >50% | >75% |
| Crystals | None | None | MSU or CPP | None (but crystals + infection can coexist) |
| Gram stain | Negative | Negative | Negative | Positive in ~50–75% |
| Culture | Negative | Negative | Negative | Positive in ~80–90% |
Corticosteroid Injection
| Joint | Agent | Dose |
|---|---|---|
| Large (knee, shoulder) | Triamcinolone acetonide or methylprednisolone acetate | 40–80 mg |
| Medium (wrist, ankle, elbow) | Triamcinolone acetonide | 20–40 mg |
| Small (MCP, PIP, MTP) | Triamcinolone acetonide | 5–10 mg |
Generally limit to 3–4 injections per joint per year. Post-injection steroid flare (within 24 hours, self-limited) occurs in ~2–5% of patients. Infection risk is ~1 in 15,000–50,000 injections.
31 Autoantibody Interpretation
ANA Patterns & Disease Associations
| Pattern | Target | Associated Disease | Notes |
|---|---|---|---|
| Homogeneous | dsDNA, histones | SLE, drug-induced lupus | Anti-histone: drug-induced lupus (95%); anti-dsDNA: active SLE, nephritis |
| Speckled | Sm, RNP, SSA/Ro, SSB/La | SLE, MCTD, Sjögren | Most common pattern; least specific |
| Nucleolar | RNA pol III, Scl-70, fibrillarin, PM-Scl | Systemic sclerosis | Nucleolar pattern strongly suggests SSc |
| Centromere | CENP-A/B/C | Limited SSc (CREST) | Distinct discrete speckled pattern |
| Peripheral (rim) | dsDNA, nuclear envelope | SLE (especially with nephritis) | Very specific for SLE |
Specific Autoantibodies
| Antibody | Disease | Clinical Significance |
|---|---|---|
| Anti-dsDNA | SLE | Specific (~95%); titers correlate with disease activity and nephritis |
| Anti-Sm (Smith) | SLE | Most specific for SLE (~99%); low sensitivity (~30%) |
| Anti-U1 RNP | MCTD; SLE | Required for MCTD diagnosis; also seen in SLE (~30%) |
| Anti-SSA/Ro | Sjögren, SLE, SCLE | Neonatal lupus + congenital heart block risk (anti-Ro52/60 crosses placenta) |
| Anti-SSB/La | Sjögren | More specific than SSA; rarely positive without SSA |
| Anti-Scl-70 | Diffuse SSc | Associated with ILD |
| Anticentromere | Limited SSc | Associated with PAH |
| Anti-RNA pol III | Diffuse SSc | Scleroderma renal crisis; malignancy |
| Anti-Jo-1 | Anti-synthetase syndrome | ILD, mechanic's hands, Raynaud, arthritis |
| Anti-histone | Drug-induced lupus | Present in ~95% of DIL; also in SLE (~70%) |
| Antiphospholipid antibodies | APS (primary or secondary to SLE) | Lupus anticoagulant, anticardiolipin, anti-B2GP1; thrombosis + pregnancy morbidity |
ANCA Testing
| Pattern | Antigen (ELISA) | Disease |
|---|---|---|
| c-ANCA | PR3 (proteinase 3) | GPA (90%) |
| p-ANCA | MPO (myeloperoxidase) | MPA, EGPA, drug-induced vasculitis |
| Atypical p-ANCA | Lactoferrin, elastase, others | IBD, autoimmune hepatitis, PSC (not true vasculitis) |
Complement Levels
Low C3 and C4: active SLE (classical pathway consumption by immune complexes). Low C4 only: cryoglobulinemia, hereditary C4 deficiency, APS. Normal or elevated complement: most other rheumatic diseases (complement is an acute-phase reactant). C3 and C4 levels are used to monitor SLE disease activity alongside anti-dsDNA titers.
32 Rheumatologic Imaging
X-Ray Findings by Disease
| Disease | Characteristic X-Ray Findings |
|---|---|
| Rheumatoid arthritis | Periarticular osteopenia, symmetric joint space narrowing, marginal erosions (MCPs, PIPs, wrists); NO osteophytes |
| Osteoarthritis | Asymmetric joint space narrowing, subchondral sclerosis, osteophytes, subchondral cysts |
| Psoriatic arthritis | Erosions WITH new bone formation (periostitis), "pencil-in-cup" deformity, ankylosis; DIP involvement |
| Ankylosing spondylitis | Bilateral symmetric sacroiliitis, syndesmophytes (marginal, vertical), "bamboo spine," "shiny corners" (Romanus lesion) |
| Gout | Punched-out erosions with overhanging edges ("rat bite"), preserved joint space (until late), tophi shadows |
| CPPD | Chondrocalcinosis (linear calcification in cartilage); knee menisci, TFCC, pubic symphysis |
| SLE (Jaccoud) | Non-erosive deformities (reducible subluxation without erosions) |
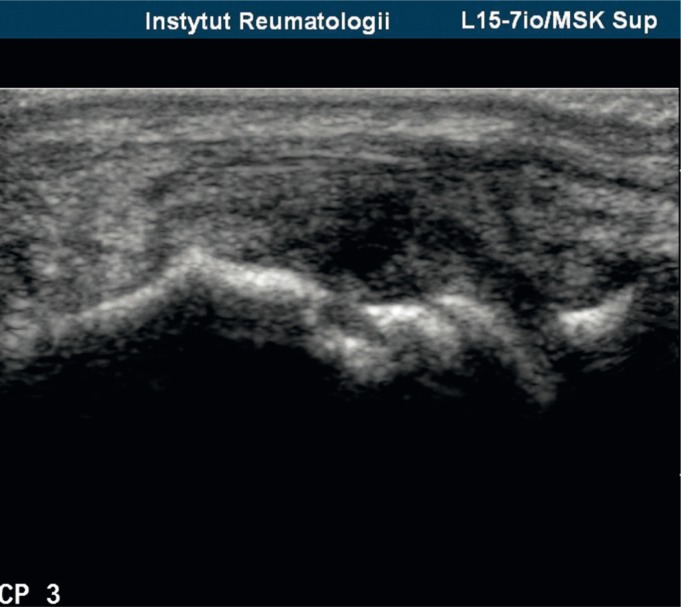
Musculoskeletal Ultrasound
Increasingly used in rheumatology for: detecting synovitis (grayscale and power Doppler), guiding joint aspiration/injection, assessing enthesitis, detecting erosions (more sensitive than X-ray for early erosions), monitoring treatment response. Advantages: no radiation, real-time, point-of-care. Limitations: operator-dependent, limited for deep structures.
MRI in Rheumatology
| Indication | MRI Findings |
|---|---|
| Early RA | Bone marrow edema (pre-erosive), synovitis with enhancement, early erosions |
| Sacroiliitis (axSpA) | Bone marrow edema at SI joints (STIR sequences) — earliest sign of axial SpA; structural changes (fat metaplasia, erosions, ankylosis) |
| Myositis | Muscle edema (STIR), fatty infiltration (chronic) |
| Osteonecrosis | "Band sign" — serpiginous low-signal line on T1 surrounding necrotic segment |
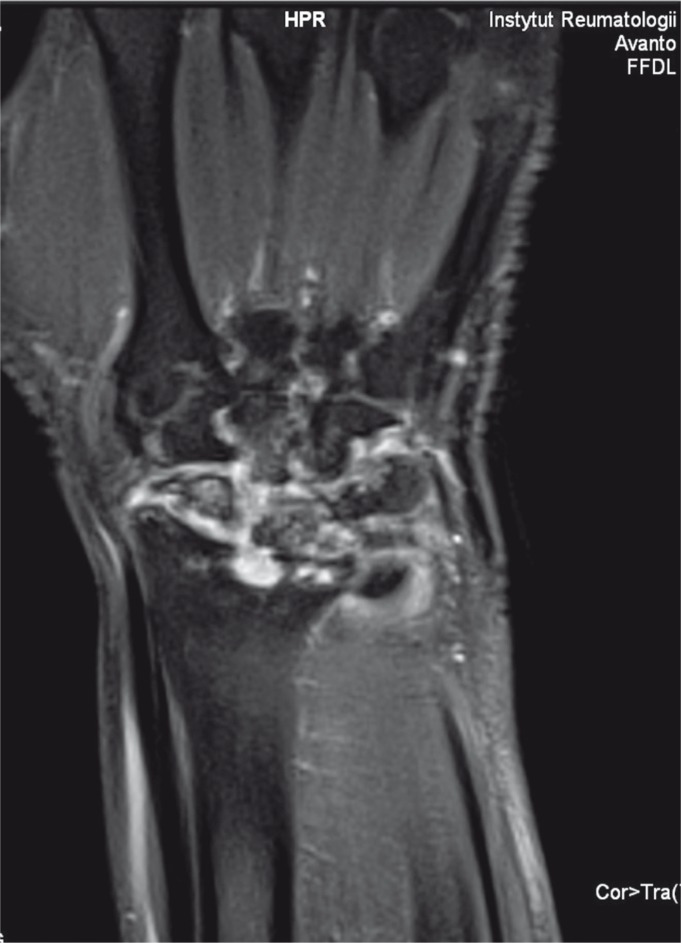
33 Imaging in Rheumatology
Imaging Modality Selection
| Clinical Question | First-Line Imaging | Second-Line / Advanced |
|---|---|---|
| Suspected RA erosions | X-ray hands and feet | MRI (early erosions, bone marrow edema); US (synovitis, power Doppler) |
| Suspected sacroiliitis | X-ray pelvis (AP view) | MRI SI joints (STIR sequences for bone marrow edema) |
| Crystal disease | X-ray (chondrocalcinosis in CPPD; erosions in gout) | Dual-energy CT (DECT) for MSU crystal detection; US ("double contour" sign in gout) |
| Vasculitis (large vessel) | CTA or MRA | PET/CT (vessel wall inflammation in GCA/Takayasu); temporal artery US |
| ILD screening (SSc, myositis) | HRCT chest | PFTs (FVC, DLCO) for serial monitoring |
| Soft tissue/tendon pathology | Musculoskeletal ultrasound | MRI (for deep structures, complex anatomy) |
| Osteonecrosis | MRI (most sensitive) | X-ray (late findings only) |
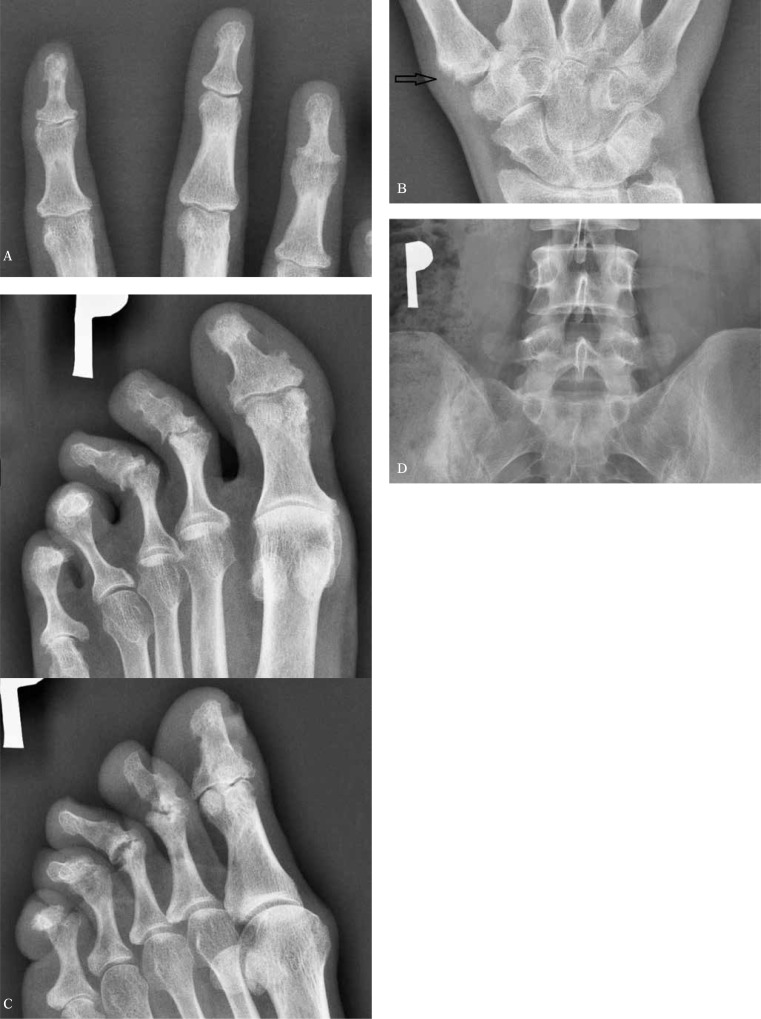
Dual-Energy CT (DECT) in Gout
DECT can identify and color-code MSU crystal deposits in joints and soft tissues with high specificity (~90–100%). Useful when joint aspiration is not feasible or results are inconclusive. Limitations: lower sensitivity in early/first-attack gout; false positives from calcium-containing deposits; expensive; radiation exposure.
34 Classification Systems & Criteria
Summary of Major Classification Criteria
| Disease | Criteria | Key Features |
|---|---|---|
| RA | 2010 ACR/EULAR | Score ≥6/10 (joint involvement, serology, acute phase reactants, duration) |
| SLE | 2019 EULAR/ACR | ANA entry criterion + score ≥10 (weighted clinical and immunologic domains) |
| PsA | CASPAR (2006) | Inflammatory articular disease + ≥3 points (psoriasis, dactylitis, nail dystrophy, juxta-articular bone, RF negative) |
| AS | Modified New York (1984) | Radiographic sacroiliitis + ≥1 clinical criterion (IBP, lumbar limitation, chest expansion) |
| Axial SpA | ASAS (2009) | Chronic back pain <45 years onset + sacroiliitis on imaging OR HLA-B27 + ≥2 SpA features |
| Vasculitis | Chapel Hill Consensus (2012) | Nomenclature system based on vessel size (large, medium, small) and pathogenesis |
| GCA | ACR 1990 | ≥3 of 5: age ≥50, new headache, temporal artery abnormality, elevated ESR, abnormal biopsy |
| GPA | ACR 1990 | ≥2 of 4: nasal/oral inflammation, abnormal CXR, urinary sediment, granulomatous inflammation on biopsy |
| SSc | 2013 ACR/EULAR | Score ≥9 (skin thickening, fingertip lesions, telangiectasia, Raynaud, SSc antibodies, PAH/ILD) |
| Sjögren | 2016 ACR/EULAR | Score ≥4 (labial gland biopsy focus score ≥1 = 3 points; anti-SSA = 3; ocular/salivary tests) |
| AOSD | Yamaguchi (1992) | ≥5 criteria (≥2 major) after exclusions; most sensitive criteria |
| Fibromyalgia | 2016 ACR revised | WPI ≥7 + SSS ≥5 OR WPI 4–6 + SSS ≥9; symptoms ≥3 months |
Chapel Hill Consensus Classification of Vasculitis (2012)
| Vessel Size | Diseases |
|---|---|
| Large vessel | Giant cell arteritis, Takayasu arteritis |
| Medium vessel | Polyarteritis nodosa, Kawasaki disease |
| Small vessel (ANCA-associated) | GPA, MPA, EGPA |
| Small vessel (immune complex) | Anti-GBM disease, IgA vasculitis, cryoglobulinemic vasculitis, hypocomplementemic urticarial vasculitis |
| Variable vessel | Behçet disease, Cogan syndrome |
| Single organ | Cutaneous leukocytoclastic vasculitis, primary CNS vasculitis |
35 Medications Master Table
Conventional Synthetic DMARDs
| Drug | Mechanism | Indications | Dose | Key Toxicities | Monitoring |
|---|---|---|---|---|---|
| Methotrexate | Folate antagonist; adenosine release | RA, PsA, GPA (maintenance), myositis, AOSD | 15–25 mg PO/SC weekly | Hepatotoxicity, cytopenias, pneumonitis, stomatitis; teratogenic | CBC, LFTs, Cr q8–12 wk; PFTs if respiratory symptoms |
| Hydroxychloroquine | Inhibits TLR signaling | SLE (all patients), RA, SS | ≤5 mg/kg/day | Retinal toxicity, QTc prolongation (rare) | Annual OCT after 5 years (or sooner if risk factors) |
| Sulfasalazine | Anti-inflammatory (NF-kB) | RA, peripheral SpA, ReA | 1–3 g/day | GI upset, rash, cytopenias, oligospermia | CBC, LFTs q8–12 wk |
| Leflunomide | Pyrimidine synthesis (DHODH) | RA, PsA | 20 mg/day | Hepatotoxicity, diarrhea, alopecia; teratogenic | CBC, LFTs monthly x6 mo then q8 wk |
| Mycophenolate | Purine synthesis (IMPDH) | LN, SSc-ILD, myositis, vasculitis maintenance | 2–3 g/day (induction); 1–2 g (maintenance) | GI (diarrhea), cytopenias, infections; teratogenic | CBC q2–4 wk initially, then q8–12 wk |
| Azathioprine | Purine synthesis; T-cell inhibition | SLE, vasculitis maintenance, myositis | 2–3 mg/kg/day | Cytopenias (check TPMT/NUDT15), hepatotoxicity, infection | TPMT genotype before starting; CBC, LFTs q4–8 wk |
| Cyclophosphamide | Alkylating agent; cytotoxic | AAV induction, severe LN, CNS lupus, SSc-ILD | IV 15 mg/kg q2–4 wk or PO 2 mg/kg/day | Cytopenias, hemorrhagic cystitis (MESNA), infections, gonadal toxicity, malignancy | CBC q2 wk; urinalysis; cumulative dose tracking |
Biologic DMARDs
| Target | Drug(s) | Route/Frequency | Key Indications | Pre-Treatment Screening |
|---|---|---|---|---|
| TNF-alpha | Etanercept, infliximab, adalimumab, golimumab, certolizumab | SC q1–4 wk or IV q4–8 wk | RA, PsA, AS, IBD (not etanercept) | TB (TST or IGRA), hepatitis B/C, exclude active infection, CHF screen |
| IL-6 receptor | Tocilizumab, sarilumab | SC q2 wk or IV q4 wk | RA, GCA, AOSD | TB, hepatitis, lipids (raises LDL), diverticulitis history |
| CD20 (B-cell) | Rituximab | IV 1000 mg x2 q6 mo | RA, AAV, myositis, SLE (off-label) | TB, hepatitis B (risk of reactivation), immunoglobulin levels |
| T-cell co-stimulation | Abatacept | SC weekly or IV q4 wk | RA, PsA | TB, hepatitis |
| BLyS (BAFF) | Belimumab | SC weekly or IV q4 wk | SLE, lupus nephritis | Standard infection screen |
| Type I IFN receptor | Anifrolumab | IV q4 wk | SLE (skin/joint dominant) | Standard infection screen; herpes zoster risk |
| IL-17A | Secukinumab, ixekizumab | SC q4 wk | PsA, AS | TB; avoid in IBD |
| IL-23 (p19) | Guselkumab, risankizumab | SC q4–8 wk | PsA | TB, infection screen |
| IL-1 | Anakinra, canakinumab | SC daily (anakinra) or q4–8 wk (canakinumab) | Gout flare, AOSD, FMF | Standard infection screen |
| C5a receptor | Avacopan | PO 30 mg BID | AAV (adjunctive, steroid-sparing) | Hepatitis B screen; LFTs |
JAK Inhibitors (Targeted Synthetic DMARDs)
| Drug | JAK Selectivity | Dose | Approved Indications | Key Safety |
|---|---|---|---|---|
| Tofacitinib | JAK1/JAK3 | 5 mg BID | RA, PsA, AS, UC | FDA boxed warning: VTE, MACE, malignancy risk (ORAL Surveillance trial in RA); herpes zoster; cytopenias; lipid elevation |
| Baricitinib | JAK1/JAK2 | 2 mg daily | RA | |
| Upadacitinib | JAK1 | 15 mg daily | RA, PsA, AS, UC, AD |
Glucocorticoid Equivalency
| Drug | Equivalent Dose (mg) | Relative Anti-Inflammatory Potency | Mineralocorticoid Activity | Duration |
|---|---|---|---|---|
| Hydrocortisone | 20 | 1 | ++ | Short (8–12h) |
| Prednisone / Prednisolone | 5 | 4 | + | Intermediate (12–36h) |
| Methylprednisolone | 4 | 5 | Minimal | Intermediate |
| Dexamethasone | 0.75 | 25–30 | None | Long (36–72h) |
36 Abbreviations Master List
37 Risk Scores & Monitoring Guidelines
Disease Activity Scores
| Score | Disease | Components | Targets |
|---|---|---|---|
| DAS28 | RA | 28 tender joint count, 28 swollen joint count, ESR (or CRP), patient global VAS | Remission <2.6; low activity 2.6–3.2; moderate 3.2–5.1; high >5.1 |
| CDAI | RA | 28 TJC + 28 SJC + patient global (0–10) + evaluator global (0–10) | Remission ≤2.8; low ≤10; moderate ≤22; high >22 |
| SDAI | RA | CDAI + CRP (mg/dL) | Remission ≤3.3 |
| SLEDAI-2K | SLE | 24 weighted items (seizure, psychosis, vasculitis, arthritis, rash, serositis, renal, hematologic, immunologic) | Remission = 0; mild ≤6; moderate 7–12; severe >12 |
| BVAS (v3) | Vasculitis | 9 organ systems, 56 items; active disease vs. persistent | BVAS = 0 indicates remission |
| BASDAI | AS / axSpA | 6 questions on fatigue, spinal pain, joint pain/swelling, enthesitis, morning stiffness severity and duration | Active disease ≥4; treatment escalation if ≥4 despite NSAIDs |
| ASDAS | AS / axSpA | Back pain, morning stiffness, patient global, peripheral joint pain/swelling + CRP | Inactive <1.3; moderate 1.3–2.1; high 2.1–3.5; very high >3.5 |
TB Screening Before Biologics
| Test | Method | Notes |
|---|---|---|
| TST (tuberculin skin test) | Intradermal PPD; read at 48–72 hours | Positive: ≥5 mm in immunosuppressed patients; may be falsely negative in immunosuppressed |
| IGRA (interferon-gamma release assay) | QuantiFERON-TB Gold or T-SPOT.TB | Preferred in BCG-vaccinated patients; not affected by BCG; single blood draw |
| If positive | Treat latent TB (isoniazid 9 months or rifampin 4 months) for ≥1–2 months BEFORE starting biologic. TNFi carries highest TB reactivation risk (especially infliximab and adalimumab > etanercept) | |
Monitoring During DMARD Therapy
| Drug | Baseline | Ongoing Monitoring |
|---|---|---|
| Methotrexate | CBC, LFTs, creatinine, hepatitis B/C, CXR | CBC, LFTs, Cr q8–12 weeks; annual hepatitis screen if risk factors |
| Hydroxychloroquine | Baseline ophthalmologic exam (OCT + visual fields) | Annual OCT after 5 years (or sooner: renal impairment, tamoxifen, high dose, >10 years of use) |
| Leflunomide | CBC, LFTs, creatinine | CBC, LFTs monthly x6 months, then q6–8 weeks; pregnancy test if applicable |
| Azathioprine | TPMT/NUDT15 genotype, CBC, LFTs | CBC q2–4 weeks initially, then q8–12 weeks; LFTs periodically |
| Mycophenolate | CBC, LFTs, pregnancy test | CBC q2–4 weeks initially, then q8–12 weeks |
| Cyclophosphamide | CBC, urinalysis, pregnancy test | CBC q2 weeks (nadir at 10–14 days); urinalysis (hemorrhagic cystitis); lifetime cumulative dose tracking |
| TNF inhibitors | TB screening, hepatitis B/C, CBC, LFTs | Annual TB screening if ongoing risk; monitor for infections |
| Tocilizumab | TB, hepatitis, CBC, LFTs, lipid panel | LFTs, CBC, lipids q4–8 weeks initially; CRP unreliable (suppressed by IL-6 blockade) |
| JAK inhibitors | TB, hepatitis, CBC, LFTs, lipids, VTE risk assessment | CBC, LFTs, lipids q8–12 weeks; lymphocyte counts; age-appropriate malignancy screening |
R References
- Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62(9):2569–2581.
- Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Arthritis Rheumatol. 2019;71(9):1400–1412.
- Fanouriakis A, Kostopoulou M, Alunno A, et al. 2019 Update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis. 2019;78(6):736–745.
- Smolen JS, Landewé RBM, Bergstra SA, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann Rheum Dis. 2023;82(1):3–18.
- FitzGerald JD, Dalbeth N, Mikuls T, et al. 2020 American College of Rheumatology guideline for management of gout. Arthritis Care Res. 2020;72(6):744–760.
- Ward MM, Deodhar A, Gensler LS, et al. 2019 Update of the American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network recommendations for the treatment of ankylosing spondylitis and nonradiographic axial spondyloarthritis. Arthritis Rheumatol. 2019;71(10):1599–1613.
- Gossec L, Baraliakos X, Kerschbaumer A, et al. EULAR recommendations for the management of psoriatic arthritis with pharmacological therapies: 2019 update. Ann Rheum Dis. 2020;79(6):700–712.
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 Revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013;65(1):1–11.
- Hellmich B, Sanchez-Alamo B, Schirmer JH, et al. EULAR recommendations for the management of ANCA-associated vasculitis: 2022 update. Ann Rheum Dis. 2024;83(1):30–47.
- Kowal-Bielecka O, Fransen J, Avouac J, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis. 2017;76(8):1327–1339.
- Rovin BH, Adler SG, Barratt J, et al. Executive summary of the KDIGO 2024 clinical practice guideline for the management of lupus nephritis. Kidney Int. 2024;105(1):31–45.
- Jayne DRW, Merkel PA, Schall TJ, Bekker P; ADVOCATE Study Group. Avacopan for the treatment of ANCA-associated vasculitis. N Engl J Med. 2021;384(7):599–609.
- Stone JH, Tuckwell K, Dimonaco S, et al. Trial of tocilizumab in giant-cell arteritis (GiACTA). N Engl J Med. 2017;377(4):317–328.
- Rovin BH, Teng YKO, Ginzler EM, et al. Efficacy and safety of voclosporin versus placebo for lupus nephritis (AURORA 1): a double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. 2021;397(10289):2070–2080.
- Lundberg IE, Tjirnstrom M, Bottai M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis. 2017;76(12):1955–1964.