Medical Genetics
Every syndrome, inheritance pattern, genetic test, therapy, abbreviation, and documentation framework you need to scribe in a medical genetics clinic.
All diagrams on this page are sourced from published educational or institutional materials rather than AI generation. Each figure caption links to the original source, and the full diagram and guideline citations are collected in the references section at the bottom.
01 Genetics Essentials — Inheritance & Molecular Foundations
Medical genetics is the specialty that evaluates, diagnoses, and manages patients with inherited or congenital disorders. A genetics clinic sees an exceptionally broad population: newborns with dysmorphic features, children with developmental delay, adults with a family history of cancer, couples seeking preconception counseling, and patients with unexplained multisystem disease. To scribe competently you must understand how genes are inherited, how variants cause disease, and how clinicians describe both.
DNA, Genes, and Chromosomes
Every human cell contains 46 chromosomes — 22 autosomal pairs and 1 sex chromosome pair (XX female, XY male). Each chromosome is a long strand of DNA wrapped around histone proteins. Genes are segments of DNA that encode proteins; humans have roughly 20,000 protein-coding genes. A variant (formerly "mutation") is any change from the reference genome. Variants can be single-nucleotide (missense, nonsense, synonymous, splice-site), small insertions/deletions (indels), copy-number variants (CNVs — large deletions or duplications), or structural rearrangements (translocations, inversions).
Patterns of Inheritance
Almost every HPI in genetics is framed by a suspected or confirmed inheritance pattern. You must be able to identify and chart these accurately.
Autosomal dominant (AD): A single pathogenic variant on one of the 22 autosomes is sufficient to cause disease. Affected individuals usually have an affected parent; each child of an affected parent has a 50% risk. Examples: Marfan, neurofibromatosis 1, Huntington, BRCA1/2, Lynch, familial hypercholesterolemia.
Autosomal recessive (AR): Two pathogenic variants (one on each allele) are required. Parents are typically unaffected carriers; each child of two carriers has a 25% risk. Examples: cystic fibrosis, sickle cell, PKU, Tay-Sachs, Wilson disease, most inborn errors of metabolism.
X-linked recessive (XLR): Males (hemizygous) are affected; females are typically carriers but can show milder disease from lyonization. Examples: Duchenne/Becker, hemophilia A/B, Fabry (often partial expression in females), fragile X (with expansion dynamics), red-green color blindness.
X-linked dominant (XLD): One X-linked variant causes disease; males are often more severely affected or lethal in utero. Examples: Rett syndrome (MECP2, usually lethal in males), X-linked hypophosphatemia, incontinentia pigmenti.
Mitochondrial: Exclusively maternal transmission — an affected mother passes the variant to all offspring, but fathers pass none. Heteroplasmy (varying proportion of mutant mtDNA) drives variable severity. Examples: MELAS, MERRF, LHON, Leigh syndrome.
Non-Mendelian Concepts You Will Hear
Penetrance — the proportion of individuals with a variant who manifest the phenotype. BRCA1 carriers have ~72% lifetime breast cancer risk (incomplete penetrance). Expressivity — the severity/range of manifestations among affected individuals. Anticipation — earlier onset and greater severity in successive generations, classically in trinucleotide repeat disorders (Huntington, myotonic dystrophy, fragile X). Imprinting — parent-of-origin-dependent gene expression (Prader-Willi vs Angelman). Uniparental disomy (UPD) — both chromosomes from one parent. Mosaicism — variant present in only a subset of cells. Locus heterogeneity — different genes causing the same phenotype (e.g., retinitis pigmentosa). Variable expressivity — especially prominent in NF1, Marfan, and tuberous sclerosis.
When the geneticist says "vertical transmission with male-to-male," they are ruling out X-linked (fathers cannot pass an X to sons) and supporting autosomal dominant. When they say "consanguinity," they are flagging elevated risk for autosomal recessive disease. Chart these framing words verbatim — they show the reasoning behind the differential.
02 Scribe Documentation Framework & Pedigree Charting
Genetics notes look different from most clinic notes. The HPI is often thinner than the family history, and the assessment frequently discusses probabilities rather than binary diagnoses. Three elements define a genetics note: the detailed three-generation pedigree, the structured dysmorphology exam, and the risk assessment / recommendation plan.
CC/Reason for referral: The clinical question driving the visit (e.g., "family history of breast cancer," "developmental delay and dysmorphic features," "elevated NIPT screen").
HPI: Pregnancy history (maternal age, exposures, prenatal screening results, complications), birth history (gestational age, mode of delivery, APGARs, birth parameters), developmental milestones, current concerns, prior genetic testing.
Family history / Pedigree: Minimum three generations. Document each relative's age, health status, ages at diagnosis, and cause and age of death. Note consanguinity, ethnic ancestry (founder effects), and recurrent miscarriages.
Physical exam: Anthropometrics with percentiles (height, weight, head circumference), dysmorphology head-to-toe, neurologic exam, and skin exam (cafe au lait, hypopigmented macules, hemangiomas).
Assessment: Differential diagnosis with inheritance pattern, recurrence risk, and probability estimates.
Plan: Testing recommended (with specific test names and lab), pre-test counseling documented, cascade testing for at-risk relatives, surveillance plan, follow-up interval.
Pedigree Symbols and Conventions
Every genetics note includes or references a pedigree. The standard NSGC pedigree nomenclature uses squares for males, circles for females, diamonds for unknown sex, filled symbols for affected individuals, a slash through the symbol for deceased, and a horizontal line between partners. Generations are labeled with Roman numerals (I, II, III) and individuals within a generation with Arabic numerals (1, 2, 3). The proband is indicated by an arrow.
Genetic counselors often draw the pedigree on paper during the visit and hand it to you at the end. Your job is to transcribe it accurately into the EHR narrative: "Paternal grandmother diagnosed with breast cancer at age 48, paternal aunt diagnosed with ovarian cancer at age 52, father unaffected at age 68." Always include age at diagnosis — it is the single most important piece of cancer family history data.
Informed Consent Documentation
Genetic testing requires documented informed consent. You will routinely chart that the clinician discussed benefits, risks, limitations, possible results (positive/pathogenic, negative, variant of uncertain significance), implications for family members, GINA (Genetic Information Nondiscrimination Act) protections, insurance implications, and the possibility of incidental/secondary findings per ACMG's ACMG SF v3.2 secondary findings list.
03 Chromosomal Aneuploidies Chromosomal
Aneuploidies — abnormal chromosome numbers — account for a large share of genetics referrals, particularly in pediatric and prenatal settings. They are most commonly caused by meiotic nondisjunction, with risk rising sharply with advanced maternal age.
Down Syndrome (Trisomy 21)
Karyotype: 47,XX,+21 or 47,XY,+21 (95% nondisjunction, 4% Robertsonian translocation, 1% mosaic). Incidence ~1 in 700 live births. Features: characteristic facies (upslanting palpebral fissures, epicanthal folds, flat nasal bridge, small ears, protruding tongue, brachycephaly), single transverse palmar crease, sandal gap toe, hypotonia, and intellectual disability. Medical comorbidities: congenital heart disease (~50%; AV septal defect most classic), duodenal atresia ("double bubble" on x-ray), Hirschsprung disease, hypothyroidism, early-onset Alzheimer disease, atlantoaxial instability, AML/ALL risk (transient myeloproliferative disorder in neonates), cataracts, and obstructive sleep apnea. Surveillance: follow the AAP health supervision guidelines with baseline echo, thyroid studies, CBC, hearing and vision screens, and periodic cervical spine and polysomnography.
Edwards Syndrome (Trisomy 18)
Karyotype: 47,+18. Incidence ~1 in 6,000. Features: clenched hands with overlapping fingers, rocker-bottom feet, microcephaly, prominent occiput, micrognathia, low-set malformed ears, congenital heart disease (VSD most common), omphalocele, and severe intellectual disability. Prognosis: historically ~90% mortality in the first year, though contemporary data reflect improved survival with proactive care.
Patau Syndrome (Trisomy 13)
Karyotype: 47,+13. Incidence ~1 in 10,000. Features: holoprosencephaly spectrum, cleft lip/palate, microphthalmia, polydactyly, cutis aplasia of the scalp, congenital heart disease, and omphalocele. Prognosis: severe; most affected children do not survive the first year.
Turner Syndrome (45,X)
Karyotype: 45,X (or mosaic 45,X/46,XX). Incidence ~1 in 2,500 live female births. Features: short stature, webbed neck, low posterior hairline, shield chest with widely spaced nipples, cubitus valgus, lymphedema of hands/feet at birth, gonadal dysgenesis with primary amenorrhea and infertility, horseshoe kidney, bicuspid aortic valve, and coarctation of the aorta. Management: growth hormone therapy, estrogen replacement, and lifelong cardiac surveillance (aortic dilation risk).
Klinefelter Syndrome (47,XXY)
Karyotype: 47,XXY. Incidence ~1 in 500–1,000 live male births. Features: tall stature, long limbs, gynecomastia, small firm testes, hypergonadotropic hypogonadism (high LH/FSH, low testosterone), infertility, learning and speech delays, and increased risk of breast cancer and osteoporosis. Management: testosterone replacement, fertility counseling, and developmental support.
Triple/quad screen and cell-free DNA (cfDNA) results appear constantly in prenatal genetics HPIs. Chart the specific risk number ("cfDNA reported a T21 risk of 1:10 with fetal fraction 8.2%") rather than "positive screen." These numbers drive the entire diagnostic pathway.
04 Microdeletion & Microduplication Syndromes Chromosomal
22q11.2 Deletion Syndrome (DiGeorge / Velocardiofacial)
Mechanism: ~3 Mb deletion on chromosome 22q11.2, usually de novo; autosomal dominant inheritance if a parent is affected. Most common microdeletion syndrome (~1 in 4,000). Remembered by the mnemonic CATCH-22: Cardiac (conotruncal defects — tetralogy of Fallot, truncus arteriosus, interrupted aortic arch), Abnormal facies, Thymic hypoplasia with T-cell immunodeficiency, Cleft palate, Hypocalcemia (from parathyroid hypoplasia), 22q11 deletion. Psychiatric illness (schizophrenia) is a major adult manifestation.
Williams Syndrome
Mechanism: 7q11.23 deletion including the ELN (elastin) gene. Features: "elfin" facies (broad forehead, stellate iris, wide mouth, full lips), supravalvular aortic stenosis, peripheral pulmonary stenosis, hypercalcemia in infancy, mild-to-moderate intellectual disability, and a characteristic "cocktail-party" overly friendly personality with preserved verbal skills.
Cri-du-chat Syndrome
Mechanism: 5p deletion. Features: high-pitched cat-like cry in infancy, microcephaly, round face, hypertelorism, intellectual disability, and cardiac defects.
Wolf-Hirschhorn Syndrome
Mechanism: 4p16.3 deletion. Features: "Greek warrior helmet" facies, severe intellectual disability, seizures, growth failure, and midline defects.
Smith-Magenis Syndrome
Mechanism: 17p11.2 deletion (RAI1). Features: brachycephaly, broad face, intellectual disability, self-injurious behavior, inverted sleep-wake cycle with nocturnal awakening.
05 Imprinting & Trinucleotide Repeat Disorders Genomic
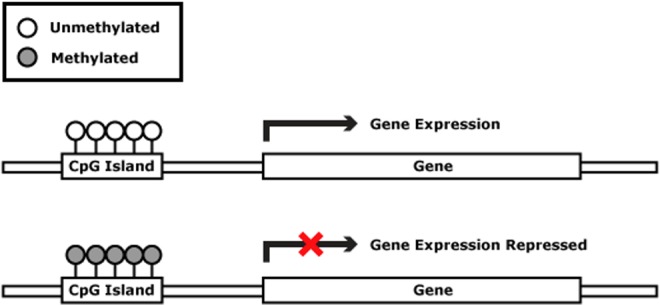
Prader-Willi Syndrome
Mechanism: Loss of paternal gene expression at 15q11-q13, from paternal deletion (~70%), maternal UPD (~25%), or imprinting defect. Features: neonatal hypotonia with poor feeding, then hyperphagia and obesity in early childhood, short stature, hypogonadism, small hands and feet, almond-shaped eyes, intellectual disability, and behavioral issues. Management: growth hormone therapy, strict diet control, behavioral support.
Angelman Syndrome
Mechanism: Loss of maternal gene expression at 15q11-q13 (UBE3A), via maternal deletion, paternal UPD, or imprinting defect. Features: severe intellectual disability, absent speech, ataxic gait ("happy puppet"), inappropriate laughter, seizures, and microcephaly.
Fragile X Syndrome
Mechanism: CGG trinucleotide repeat expansion in the FMR1 gene on Xq27.3. Normal < 45 repeats; premutation 55–200; full mutation > 200 with hypermethylation and gene silencing. X-linked; males more severely affected. Most common inherited cause of intellectual disability. Features: long narrow face, large protruding ears, macroorchidism post-puberty, intellectual disability, hyperactivity, autism spectrum features, and joint hypermobility. Premutation carriers are at risk for FXTAS (fragile X-associated tremor/ataxia syndrome) and FXPOI (primary ovarian insufficiency).
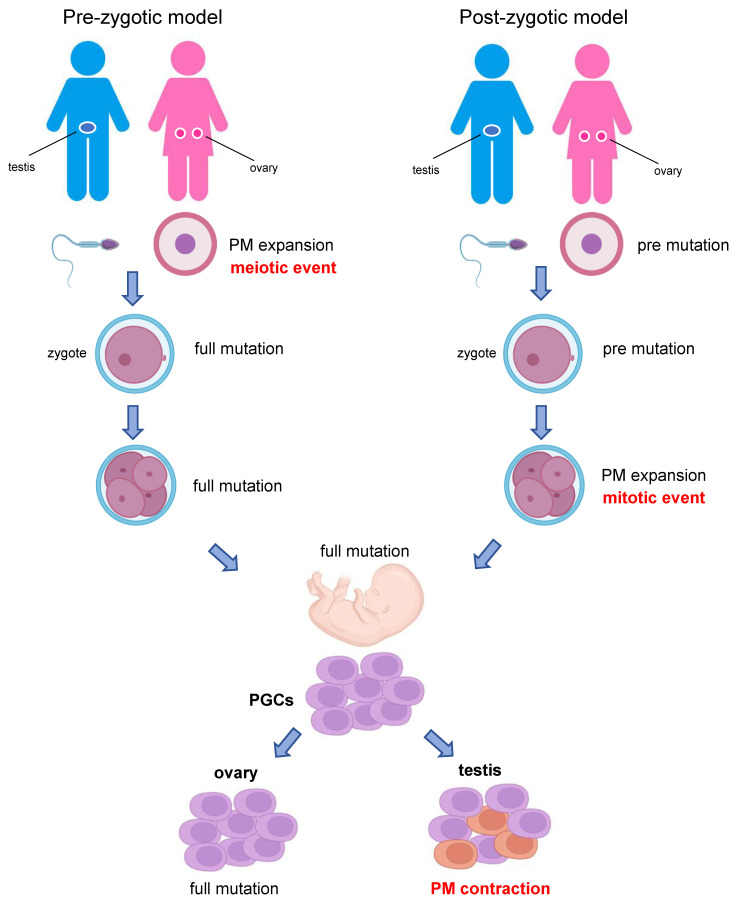
Huntington Disease
Mechanism: CAG repeat expansion in HTT on chromosome 4. Normal < 27; intermediate 27–35; reduced penetrance 36–39; full penetrance ≥ 40. Autosomal dominant with complete penetrance above 40 repeats and strong anticipation (paternal transmission). Features: adult-onset (30s–50s) chorea, dystonia, personality change, psychiatric symptoms, and progressive dementia. Imaging shows caudate atrophy. Predictive testing requires structured counseling per HDSA guidelines.
Myotonic Dystrophy Type 1
Mechanism: CTG repeat expansion in DMPK. AD with prominent anticipation; congenital form (> 1,000 repeats) passed from an affected mother. Features: myotonia (delayed muscle relaxation — grip myotonia on exam), distal weakness, frontal balding, cataracts, cardiac conduction defects, insulin resistance, and testicular atrophy.
Friedreich Ataxia
Mechanism: GAA repeat expansion in FXN (frataxin), autosomal recessive. Features: progressive gait and limb ataxia, dysarthria, loss of position/vibration sense, areflexia, pes cavus, scoliosis, hypertrophic cardiomyopathy, and diabetes.
06 Connective Tissue & Skeletal Dysplasias Structural
Marfan Syndrome
Gene: FBN1 (fibrillin-1), 15q21.1, AD. Features (revised Ghent criteria): aortic root dilation/dissection, ectopia lentis, tall stature with long limbs and arachnodactyly (positive wrist and thumb signs), pectus deformity, scoliosis, high-arched palate, skin striae, and dural ectasia. Management: beta-blockers or losartan for aortic protection, annual echocardiography, prophylactic aortic root replacement when root ≥ 5.0 cm (sooner with family history of dissection). Cite the 2022 ACC/AHA thoracic aortic disease guideline for surgical thresholds.
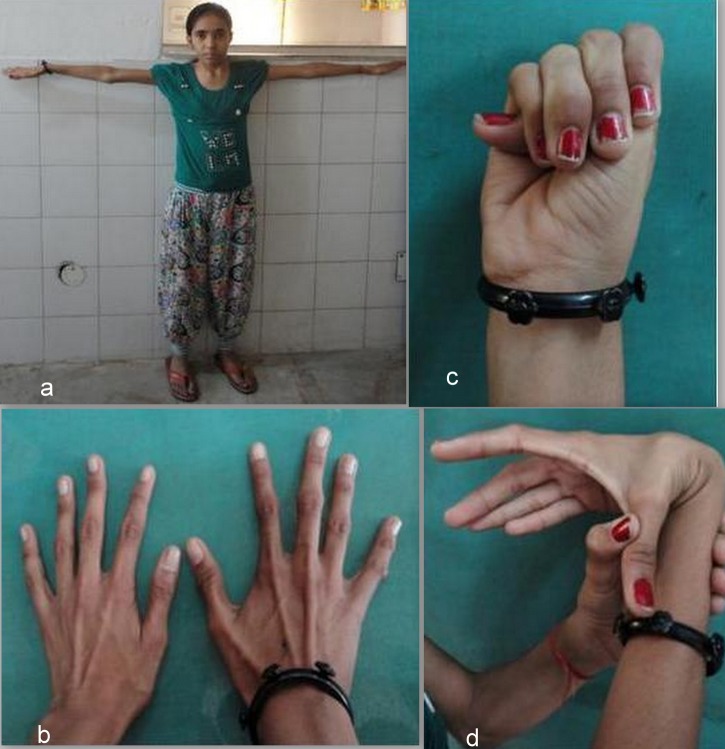
Ehlers-Danlos Syndromes (EDS)
A family of heritable connective tissue disorders defined by the 2017 international classification.
| Type | Gene | Inheritance | Key Features |
|---|---|---|---|
| Classical (cEDS) | COL5A1/A2 | AD | Skin hyperextensibility, atrophic "cigarette-paper" scars, joint hypermobility |
| Hypermobile (hEDS) | Unknown | AD | Generalized joint hypermobility, chronic pain, soft skin (clinical diagnosis per 2017 criteria) |
| Vascular (vEDS) | COL3A1 | AD | Thin translucent skin, arterial/intestinal/uterine rupture, characteristic facies |
| Kyphoscoliotic | PLOD1 | AR | Congenital kyphoscoliosis, muscle hypotonia, scleral fragility |
Vascular EDS is a medical emergency phenotype — affected patients may present with spontaneous arterial dissection at a young age and warrant celiprolol and lifelong vascular surveillance.
Loeys-Dietz Syndrome
Genes: TGFBR1, TGFBR2, SMAD3, TGFB2, TGFB3 (all AD). Features: aggressive aortic aneurysms with high dissection risk at smaller diameters than Marfan, arterial tortuosity, hypertelorism, bifid uvula or cleft palate, and craniosynostosis. Surgical thresholds are lower (often 4.0–4.5 cm at the root).
Osteogenesis Imperfecta
Genes: COL1A1, COL1A2 (most; AD). Features: recurrent fractures with minimal trauma, blue sclerae, dentinogenesis imperfecta, conductive hearing loss, short stature, and joint hypermobility. Sillence classification Types I (mild), II (perinatal lethal), III (progressive deforming), and IV (moderate). Bisphosphonates reduce fracture rate.
Achondroplasia
Gene: FGFR3 activating variant (c.1138G>A, p.Gly380Arg in >98%). AD; >80% de novo, associated with advanced paternal age. Features: disproportionate short stature (rhizomelic shortening), macrocephaly with frontal bossing, trident hand, lumbar hyperlordosis, and foramen magnum stenosis. Vosoritide (CNP analog) is an approved growth-promoting therapy.
07 Neurocutaneous & Tumor Predisposition Syndromes Tumor Risk
Neurofibromatosis Type 1 (NF1)
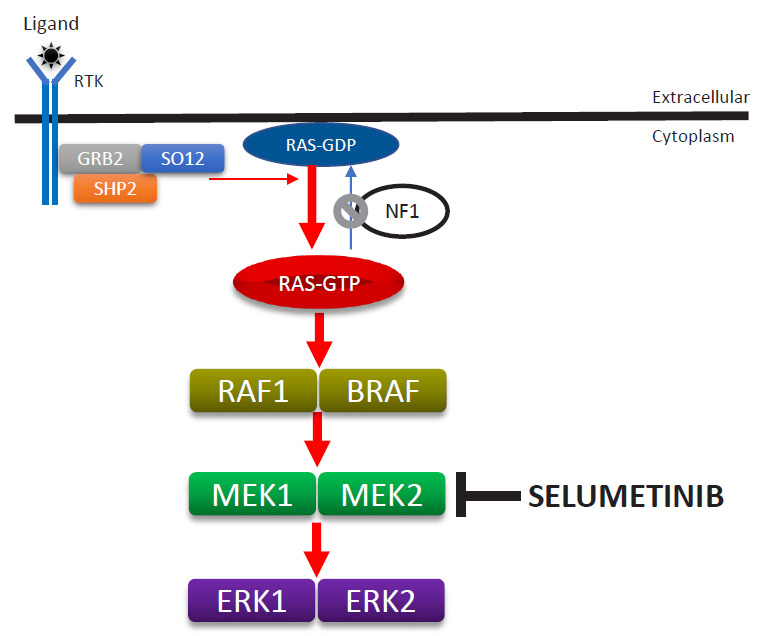
Gene: NF1 (neurofibromin), 17q11.2, AD, near-complete penetrance with variable expressivity. NIH diagnostic criteria (need 2+): ≥ 6 cafe-au-lait macules (≥ 5 mm prepubertal, ≥ 15 mm postpubertal), ≥ 2 neurofibromas or 1 plexiform neurofibroma, axillary/inguinal freckling (Crowe sign), optic pathway glioma, ≥ 2 Lisch nodules (iris hamartomas), distinctive osseous lesion (sphenoid dysplasia, tibial pseudarthrosis), or affected first-degree relative. Selumetinib (MEK inhibitor) is approved for inoperable plexiform neurofibromas.
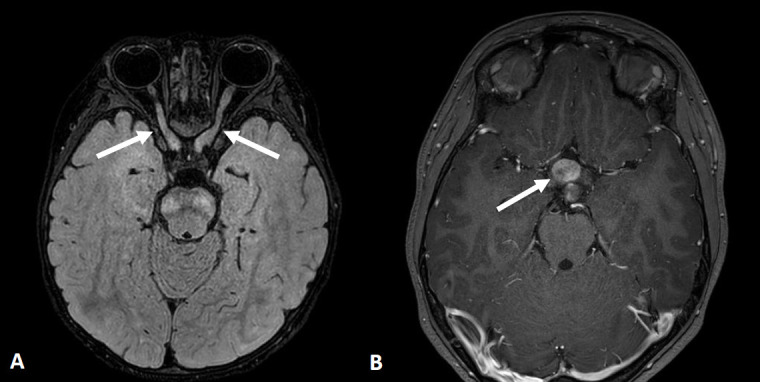
Neurofibromatosis Type 2 (NF2)
Gene: NF2 (merlin), 22q12.2, AD. Features: bilateral vestibular schwannomas (hallmark), meningiomas, ependymomas, juvenile cataracts. Presents with sensorineural hearing loss and tinnitus.
Tuberous Sclerosis Complex (TSC)
Genes: TSC1 (hamartin) or TSC2 (tuberin), AD. Features: cortical tubers, subependymal nodules, SEGAs, facial angiofibromas, ash-leaf hypomelanotic macules, shagreen patches, ungual fibromas, cardiac rhabdomyomas, renal angiomyolipomas, and lymphangioleiomyomatosis (LAM). Infantile spasms and autism are common. mTOR inhibitors (everolimus, sirolimus) treat SEGAs and AMLs.
Von Hippel-Lindau (VHL)
Gene: VHL, 3p25, AD tumor suppressor. Tumors: cerebellar and retinal hemangioblastomas, clear-cell renal cell carcinoma, pheochromocytoma, pancreatic neuroendocrine tumors, endolymphatic sac tumors. Belzutifan (HIF-2α inhibitor) is approved for VHL-associated tumors.
Peutz-Jeghers Syndrome
Gene: STK11 (LKB1), AD. Features: mucocutaneous pigmentation of lips/buccal mucosa, hamartomatous GI polyps with intussusception risk, and elevated risk of GI, breast, pancreatic, and gynecologic cancers.
Cowden Syndrome / PTEN Hamartoma Tumor Syndrome
Gene: PTEN, AD. Macrocephaly, trichilemmomas, oral papillomas, Lhermitte-Duclos, and elevated risk of breast, thyroid (follicular), endometrial, and renal cancers.
Juvenile Polyposis Syndrome
Genes: SMAD4, BMPR1A, AD. Multiple juvenile hamartomatous polyps with colorectal and upper GI cancer risk. SMAD4 carriers also have HHT (hereditary hemorrhagic telangiectasia) overlap.
08 Hereditary Cancer Syndromes Cancer
Adult cancer genetics is a major portion of most clinics. Testing and surveillance are governed by the NCCN Genetic/Familial High-Risk Assessment: Breast, Ovarian, Pancreatic and NCCN Colorectal Genetic/Familial High-Risk guidelines, which a scribe will see referenced constantly.
Hereditary Breast and Ovarian Cancer (HBOC) — BRCA1/BRCA2
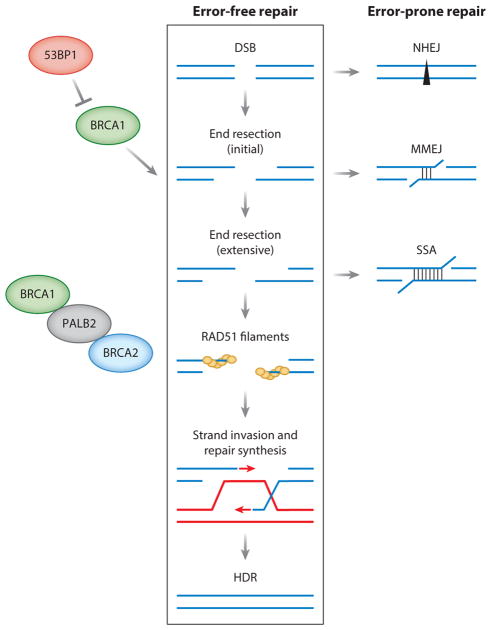
Genes: BRCA1 (17q21), BRCA2 (13q12), AD tumor suppressors. Cancer risks: BRCA1 ~65–72% lifetime breast cancer, 39–44% ovarian, elevated triple-negative and contralateral risk. BRCA2 ~69% breast, 17% ovarian, increased male breast, prostate, and pancreatic cancer. Surveillance: annual breast MRI + mammogram starting at age 25–30, risk-reducing bilateral salpingo-oophorectomy at 35–40 (BRCA1) or 40–45 (BRCA2), consider risk-reducing mastectomy. PARP inhibitors (olaparib, talazoparib) treat BRCA-associated cancers.
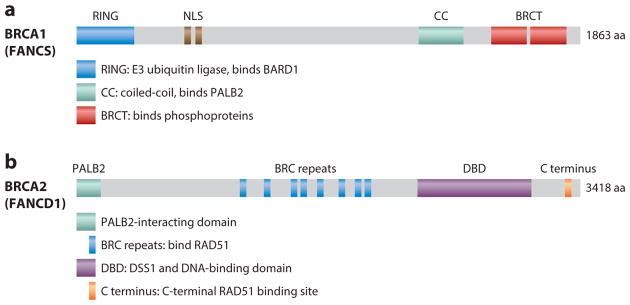
Li-Fraumeni Syndrome
Gene: TP53, AD. Core tumors: soft tissue sarcoma, osteosarcoma, premenopausal breast cancer, brain tumors, adrenocortical carcinoma, leukemia. Extremely high penetrance. Surveillance uses the "Toronto protocol" with annual whole-body MRI, brain MRI, breast MRI, and abdominal ultrasound. Avoid radiation when possible.
Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer)
Genes: MLH1, MSH2, MSH6, PMS2, EPCAM (AD mismatch repair defects). Cancer risks: colorectal (up to 80%), endometrial (up to 60%), ovarian, gastric, small bowel, urothelial, and sebaceous neoplasms (Muir-Torre variant). Tumor screening: universal MMR IHC and/or MSI testing on colorectal and endometrial cancers per NCCN and universal Lynch screening recommendations. Surveillance: colonoscopy every 1–2 years starting age 20–25, consider risk-reducing hysterectomy/BSO after childbearing.
Familial Adenomatous Polyposis (FAP)
Gene: APC, 5q, AD. Hundreds to thousands of colorectal adenomas with near-100% colorectal cancer risk if untreated; also duodenal/ampullary cancer, desmoid tumors, osteomas (Gardner variant), and hepatoblastoma in children. Management: prophylactic colectomy, typically in late teens to early 20s, with ongoing upper GI surveillance.
MYH-Associated Polyposis (MAP)
Gene: MUTYH, AR (biallelic). Attenuated polyposis phenotype. Surveillance colonoscopy every 1–2 years from age 25–30.
Hereditary Diffuse Gastric Cancer
Gene: CDH1 (E-cadherin), AD. Very high diffuse (signet-ring) gastric cancer risk; lobular breast cancer risk in females. Prophylactic total gastrectomy is typically recommended by age 20–30.
Other Hereditary Cancer Genes
Other genes routinely tested on multigene panels include PALB2, CHEK2, ATM, RAD51C/D, BARD1, BRIP1 (breast/ovarian moderate-to-high risk), NF1 (breast), and STK11/PTEN (syndromic). Variant reporting follows ACMG criteria.
09 Multiple Endocrine Neoplasia & Endocrine Genetics Endocrine
MEN1 (Wermer Syndrome)
Gene: MEN1 (menin), AD. Triad — "3 Ps": Parathyroid hyperplasia/adenomas (most common, earliest), Pancreatic neuroendocrine tumors (gastrinoma, insulinoma), Pituitary adenomas (prolactinoma most common). Foregut carcinoids and adrenal adenomas also occur.
MEN2A (Sipple Syndrome)
Gene: RET proto-oncogene, AD. Features: medullary thyroid carcinoma (nearly 100%), pheochromocytoma (~50%), parathyroid hyperplasia (~25%). Prophylactic thyroidectomy is recommended in childhood based on specific RET codon variant risk stratification.
MEN2B
Gene: RET (M918T in >95%), AD. Features: very early-onset aggressive MTC, pheochromocytoma, mucosal neuromas, marfanoid habitus, intestinal ganglioneuromatosis. Prophylactic thyroidectomy is recommended in the first year of life.
Familial Isolated Pheochromocytoma/Paraganglioma
SDHA/B/C/D, SDHAF2, MAX, TMEM127, VHL, RET, NF1 — most pheochromocytomas have an underlying germline variant, so genetic testing is recommended for nearly all patients.
Congenital Adrenal Hyperplasia (CAH)
Gene: CYP21A2 (21-hydroxylase deficiency accounts for >95%), AR. Classic salt-wasting form presents with neonatal adrenal crisis, ambiguous genitalia in females, and elevated 17-hydroxyprogesterone. Non-classic form presents with hirsutism, oligomenorrhea, and precocious puberty. Treatment: glucocorticoid ± mineralocorticoid replacement. Included in newborn screening.
10 Inborn Errors of Metabolism Metabolic
Inborn errors of metabolism (IEM) are a broad category of typically autosomal recessive enzyme deficiencies. Many present in the neonatal period with encephalopathy, vomiting, hypoglycemia, acidosis, or hyperammonemia. The ACMG Recommended Uniform Screening Panel (RUSP) guides newborn screening and is referenced in almost every neonatal genetics HPI.
Amino Acid Disorders
Phenylketonuria (PKU): PAH deficiency, AR. Untreated → intellectual disability, seizures, eczema, musty body odor, hypopigmentation. Newborn screening is universal. Lifelong phenylalanine-restricted diet; sapropterin (Kuvan) in responders; pegvaliase for adults. Pregnant PKU patients must maintain strict metabolic control to prevent maternal PKU embryopathy.
Maple Syrup Urine Disease (MSUD): BCKDH deficiency, AR. Elevated branched-chain amino acids (leucine, isoleucine, valine); sweet-smelling urine, neonatal encephalopathy. Treated with BCAA-restricted diet; liver transplant in severe cases.
Homocystinuria: CBS deficiency, AR. Marfanoid habitus, lens dislocation (downward), thromboembolism, intellectual disability. Treated with B6, betaine, folate, B12, and methionine restriction.
Tyrosinemia Type 1: FAH deficiency, AR. Liver failure, renal tubulopathy, hepatocellular carcinoma risk. Nitisinone (NTBC) plus tyrosine/phenylalanine-restricted diet.
Urea Cycle Disorders
OTC deficiency (X-linked, most common), CPS1, ASS1 (citrullinemia), ASL, ARG1. Present with hyperammonemia (neonatal or later), lethargy, vomiting, and encephalopathy. Treatment: protein restriction, nitrogen scavengers (sodium phenylbutyrate, sodium benzoate, glycerol phenylbutyrate — Ravicti), arginine/citrulline supplementation, hemodialysis for acute crises, and liver transplant.
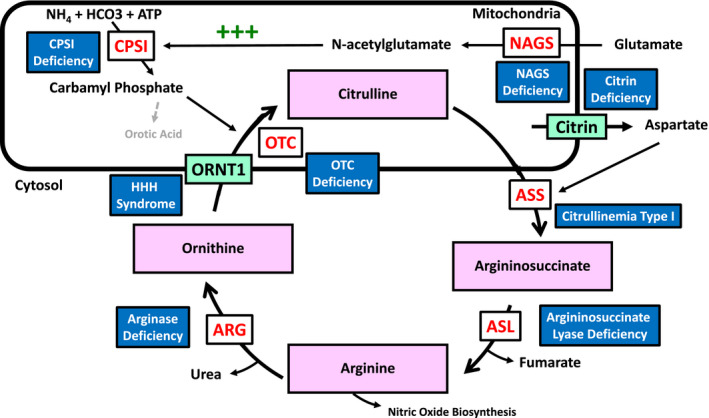
Organic Acidemias
Methylmalonic acidemia, propionic acidemia, isovaleric acidemia, glutaric acidemia type 1. Present with metabolic acidosis, hyperammonemia, and ketosis. Managed with protein restriction, carnitine, cofactor supplementation, and emergency protocols.
Fatty Acid Oxidation Disorders
MCAD deficiency is the most common and is on newborn screening. Presents with fasting hypoglycemia, Reye-like syndrome. Treated with avoidance of fasting. VLCAD and LCHAD are more severe and include cardiomyopathy and rhabdomyolysis.
Neonatal metabolic HPIs are dense with numbers — plasma ammonia, lactate, pH, bicarbonate, anion gap, glucose, ketones, acylcarnitine profile, urine organic acids, plasma amino acids. Chart every number. The geneticist's assessment depends on the specific pattern and magnitude of abnormalities.
11 Lysosomal Storage & Glycogen Storage Disorders Metabolic
Gaucher Disease
Gene: GBA (glucocerebrosidase), AR. Type 1 (non-neuronopathic — most common, Ashkenazi Jewish founder effect) — hepatosplenomegaly, cytopenias, bone pain, avascular necrosis. Type 2 (acute neuronopathic, infantile). Type 3 (chronic neuronopathic). Treatment: enzyme replacement therapy (ERT) with imiglucerase (Cerezyme), velaglucerase, taliglucerase; substrate reduction therapy (SRT) with eliglustat or miglustat.
Fabry Disease
Gene: GLA (alpha-galactosidase A), X-linked. Accumulation of globotriaosylceramide. Features: neuropathic pain (acroparesthesias), angiokeratomas, cornea verticillata, hypohidrosis, progressive renal disease, cardiomyopathy, early strokes. Treatment: ERT with agalsidase beta (Fabrazyme), agalsidase alfa; oral chaperone migalastat (Galafold) in amenable variants.
Mucopolysaccharidoses (MPS)
MPS I (Hurler/Scheie) — IDUA: coarse facies, dysostosis multiplex, corneal clouding, hepatosplenomegaly. Treated with laronidase (Aldurazyme) and hematopoietic stem cell transplant in severe forms. MPS II (Hunter) — IDS, X-linked: idursulfase (Elaprase). MPS IV (Morquio) — elosulfase alfa. MPS VI — galsulfase.
Pompe Disease
Gene: GAA, AR. Glycogen storage disease type II; both lysosomal and glycogen storage disorder. Infantile form: severe hypertrophic cardiomyopathy, hypotonia, death without treatment. Late-onset: progressive proximal weakness, respiratory failure. Treatment: alglucosidase alfa (Myozyme/Lumizyme), avalglucosidase alfa (Nexviazyme).
Tay-Sachs Disease
Gene: HEXA, AR. Ashkenazi Jewish and French Canadian founder populations. Progressive neurodegeneration in infancy with cherry-red macula, hyperacusis, and hypotonia; death by age 4–5. No disease-modifying therapy.
Niemann-Pick Disease
Type A/B (SMPD1 — acid sphingomyelinase, AR); Type C (NPC1/NPC2 — cholesterol trafficking, AR). Type C treated with miglustat.
Glycogen Storage Diseases
Von Gierke (GSD I) — G6PC: severe fasting hypoglycemia, hepatomegaly, doll-like facies, lactic acidosis, hyperuricemia. Managed with frequent feeds and cornstarch. Pompe (GSD II) — see above. McArdle (GSD V) — PYGM: exercise intolerance, "second wind" phenomenon, myoglobinuria with exercise.
12 Mitochondrial Disorders Metabolic
Mitochondrial disorders can be caused by variants in either mitochondrial DNA (mtDNA, maternal inheritance, heteroplasmy) or nuclear-encoded mitochondrial proteins (Mendelian inheritance). They classically affect high-energy tissues: brain, muscle, eye, heart, and endocrine.
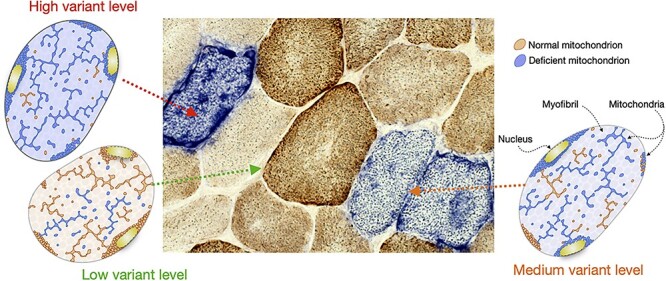
MELAS
Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes. Most common variant: m.3243A>G in MT-TL1. Features: stroke-like episodes not confined to vascular territories, seizures, sensorineural hearing loss, short stature, diabetes, lactic acidosis. Treated supportively; L-arginine during acute stroke-like episodes.
MERRF
Myoclonic Epilepsy with Ragged-Red Fibers. m.8344A>G in MT-TK. Features: myoclonus, epilepsy, ataxia, myopathy with ragged-red fibers on muscle biopsy.
Leigh Syndrome
Subacute necrotizing encephalomyelopathy; many nuclear and mtDNA causes. Features: psychomotor regression, brainstem and basal ganglia lesions on MRI, lactic acidosis, respiratory failure.
LHON
Leber Hereditary Optic Neuropathy. m.11778, m.3460, m.14484. Subacute bilateral painless central vision loss in young men. Idebenone is an approved treatment.
Kearns-Sayre Syndrome
Single large mtDNA deletion. Triad: progressive external ophthalmoplegia, pigmentary retinopathy, onset before age 20. Cardiac conduction block, cerebellar ataxia, and elevated CSF protein are common.
13 Hematologic Genetic Disorders & Hypercoagulable States Hematologic
Sickle Cell Disease
Gene: HBB, AR. Homozygous HbSS or compound heterozygotes (HbSC, HbS/beta-thal). Glu6Val substitution in beta-globin causes polymerization of deoxyhemoglobin. Features: vaso-occlusive crises, acute chest syndrome, stroke, splenic sequestration, osteonecrosis, dactylitis. Treatment: hydroxyurea, L-glutamine, crizanlizumab, voxelotor, chronic transfusion, HSCT, and newer gene therapies (exa-cel, lovo-cel). Universal newborn screening.
Thalassemia
Alpha-thalassemia: HBA1/HBA2 deletions. Silent carrier (1 deletion), trait (2), HbH disease (3), Bart hydrops (4, typically lethal in utero). Beta-thalassemia: HBB point variants. Minor (trait), intermedia, major (Cooley anemia) — transfusion-dependent anemia with skeletal changes, iron overload requiring chelation (deferasirox, deferoxamine, deferiprone). Luspatercept and gene therapy (beti-cel) are newer options.
Hemophilia A & B
Gene: F8 (A) or F9 (B), X-linked recessive. Features: spontaneous joint and soft tissue bleeding; severity correlates with factor level (severe < 1%, moderate 1–5%, mild 5–40%). Treated with factor concentrate (recombinant FVIII/FIX), emicizumab (bispecific antibody for hemophilia A), and gene therapy (etranacogene dezaparvovec for hemophilia B, valoctocogene for A).
Von Willebrand Disease
Most common inherited bleeding disorder. Types 1 (quantitative, AD), 2 (qualitative, subtypes 2A/2B/2M/2N), 3 (severe quantitative, AR). Mucocutaneous bleeding, menorrhagia, epistaxis. Treated with desmopressin, VWF concentrates, tranexamic acid.
Inherited Thrombophilias
Factor V Leiden: F5 G1691A — activated protein C resistance; most common inherited thrombophilia (~5% of Europeans). Prothrombin G20210A: F2 variant increasing prothrombin levels. Antithrombin, protein C, protein S deficiencies: rarer but stronger thrombotic risk. MTHFR C677T/A1298C: mild hyperhomocysteinemia; current ACMG and ACMG practice guidance recommends against routine MTHFR testing for thrombophilia given minimal clinical utility. Hypercoagulable workup is generally reserved for unprovoked VTE, unusual sites, or strong family history.
14 Neuromuscular & Neurogenetic Disorders Neurologic
Duchenne / Becker Muscular Dystrophy
Gene: DMD (dystrophin), Xp21, X-linked recessive. Duchenne — out-of-frame deletions/duplications with absent dystrophin; onset ages 2–5, Gowers sign, calf pseudohypertrophy, loss of ambulation by early teens, dilated cardiomyopathy, respiratory failure. Becker — in-frame variants with reduced dystrophin; milder and later onset. Treatment: corticosteroids (prednisone, deflazacort, vamorolone), exon-skipping antisense oligonucleotides (eteplirsen for exon 51, golodirsen/viltolarsen for exon 53, casimersen for exon 45), ataluren (stop-codon readthrough), and delandistrogene moxeparvovec (Elevidys) gene therapy.
Spinal Muscular Atrophy (SMA)
Gene: SMN1, AR (homozygous exon 7 deletion in ~95%); SMN2 copy number modifies severity. Types 0–4 range from prenatal onset to adult. Treatments: nusinersen (Spinraza) intrathecal antisense oligonucleotide, onasemnogene abeparvovec (Zolgensma) single-dose gene therapy for children < 2, and risdiplam (Evrysdi) oral SMN2 splicing modifier. Added to RUSP in 2018, dramatically improving outcomes.
Charcot-Marie-Tooth (CMT)
Heterogeneous inherited peripheral neuropathies. CMT1A (PMP22 duplication, AD, most common) — demyelinating, distal weakness, foot drop, pes cavus, hammer toes, "stork leg" deformity. CMT2 (axonal), CMTX (GJB1), and many other subtypes.
Rett Syndrome
Gene: MECP2, X-linked dominant. Almost exclusively affects girls (lethal in most males). Features: normal development to ~6–18 months, then regression with loss of purposeful hand use, stereotypic hand-wringing, gait apraxia, acquired microcephaly, seizures, and breathing irregularities. Trofinetide is an approved disease-modifying therapy.
ALS (Familial)
~10% of ALS is familial. SOD1 (first identified), C9orf72 (hexanucleotide repeat, most common, AD), TARDBP, FUS. Tofersen (SOD1 antisense oligonucleotide) is approved for SOD1-ALS.
Autism Spectrum Disorder Genetics
ASD has strong genetic contributions. Chromosomal microarray (CMA) and fragile X testing are first-tier per ACMG; exome sequencing is recommended as second-tier or even first-tier per updated guidance. Syndromic causes include fragile X, Rett, tuberous sclerosis, Angelman, Smith-Lemli-Opitz, and Phelan-McDermid (22q13 deletion).
Congenital Heart Disease Genetics
Many CHDs have a genetic basis: 22q11.2 deletion (conotruncal), Williams (supravalvular AS), Noonan (pulmonary stenosis, HCM — PTPN11, RAF1, SOS1, RASopathies), Alagille (JAG1, NOTCH2 — peripheral pulmonary stenosis, butterfly vertebrae, posterior embryotoxon, cholestasis), Holt-Oram (TBX5 — ASD plus upper limb anomalies).
15 Cystic Fibrosis & Congenital Multisystem Disorders Multisystem
Cystic Fibrosis
Gene: CFTR, 7q31.2, AR. F508del accounts for ~70% of CF alleles in individuals of European ancestry. Features: chronic sinopulmonary disease with Pseudomonas colonization, pancreatic insufficiency, meconium ileus in neonates, male infertility from congenital bilateral absence of the vas deferens (CBAVD), CF-related diabetes, and liver disease. Diagnosed by elevated sweat chloride (≥ 60 mmol/L), positive newborn screen (IRT), or two pathogenic CFTR variants. Treatment: CFTR modulators are transformative — ivacaftor (Kalydeco) for gating variants, lumacaftor/ivacaftor (Orkambi), tezacaftor/ivacaftor (Symdeko), and elexacaftor/tezacaftor/ivacaftor (Trikafta) for ≥ 90% of CF patients. Also: airway clearance, inhaled antibiotics (tobramycin, aztreonam), dornase alfa, hypertonic saline, pancreatic enzyme replacement, fat-soluble vitamins. Preconception carrier screening for CF is standard per ACMG carrier screening recommendations.
Alpha-1 Antitrypsin Deficiency
Gene: SERPINA1, codominant. PiMM normal; PiZZ severe deficiency → early-onset panacinar emphysema and liver disease. Treatment: augmentation therapy with pooled human alpha-1 antitrypsin (Prolastin, Aralast, Zemaira, Glassia), smoking cessation, and liver transplant for end-stage liver disease.
Hereditary Hemochromatosis
Gene: HFE (C282Y homozygous most common), AR. Iron overload with cirrhosis, diabetes ("bronze diabetes"), cardiomyopathy, arthropathy, hypogonadism. Treated with therapeutic phlebotomy.
Wilson Disease
Gene: ATP7B, AR. Copper accumulation. Features: liver disease, neuropsychiatric symptoms (tremor, dystonia, personality change), Kayser-Fleischer rings. Low ceruloplasmin, elevated urinary copper. Treated with penicillamine, trientine, or zinc.
Hereditary ATTR Amyloidosis
Gene: TTR, AD. Variant transthyretin misfolds into amyloid. Features: progressive sensorimotor polyneuropathy, autonomic dysfunction, and/or restrictive cardiomyopathy. Treatments: tafamidis (stabilizer), patisiran (Onpattro, siRNA), vutrisiran (Amvuttra, siRNA), and inotersen (Tegsedi, antisense).
16 Genetic Testing & Procedures Diagnostics
A genetics clinician chooses among several tiers of testing depending on the clinical question. You will document which test was ordered, the lab, and the pre-test counseling provided.
| Test | What It Detects | Typical Use |
|---|---|---|
| Karyotype | Numerical and large (> 5–10 Mb) structural chromosome abnormalities | Suspected aneuploidy, balanced translocation, recurrent pregnancy loss |
| FISH | Specific locus or chromosome using fluorescent probes | Rapid aneuploidy detection; targeted microdeletion (22q11.2) |
| Chromosomal Microarray (CMA) | Copy number variants > ~50 kb; SNP arrays also detect UPD and ROH | First-tier test for unexplained DD/ID, ASD, MCA |
| Single-gene sequencing | SNVs/indels in one gene | Strong clinical suspicion of a specific condition |
| Multigene panel | Dozens to hundreds of genes for a phenotype | Hereditary cancer, cardiomyopathy, epilepsy, neuropathy |
| Exome sequencing (ES/WES) | Protein-coding regions of ~20,000 genes | Broad phenotype, prior testing non-diagnostic, critical NICU cases |
| Genome sequencing (GS/WGS) | Coding + noncoding + structural variants | Advanced diagnostic odyssey; rapid genome in NICU |
| Methylation studies | Imprinting status | Prader-Willi/Angelman, Beckwith-Wiedemann, Russell-Silver |
| MLPA | Single or multi-exon deletions/duplications | DMD, SMN1, hereditary cancer CNVs |
| Mitochondrial DNA sequencing | mtDNA variants, deletions, heteroplasmy | Suspected mitochondrial disease (blood, muscle, or urine) |
| Biochemical testing | Enzymes, metabolites, acylcarnitines | IEM screening (plasma amino acids, urine organic acids, lactate, ammonia) |
| Repeat expansion testing | Trinucleotide/tetranucleotide repeats | Fragile X, Huntington, myotonic dystrophy, Friedreich |
ACMG/AAP guidance establishes chromosomal microarray as the first-tier cytogenetic test for patients with unexplained developmental delay/intellectual disability, autism, and multiple congenital anomalies, replacing the older standard of G-banded karyotype for that indication.
Test names have a lot of synonyms. "CMA," "chromosomal microarray," "SNP array," and "aCGH" all refer to the same category of test. "Exome" and "WES" are interchangeable. When the clinician says "we'll start with a 84-gene cardiomyopathy panel at Invitae" — chart the specific panel name, gene count, and laboratory verbatim.
17 Prenatal Diagnosis & Reproductive Genetics Prenatal
Prenatal Screening
First-trimester combined screen (10–13+6 weeks): nuchal translucency + PAPP-A + free beta-hCG. Quad screen (15–22 weeks): AFP, hCG, uE3, inhibin A. Integrated/sequential combines first- and second-trimester markers. Cell-free DNA (NIPT/NIPS): sequences placental-origin cfDNA from maternal blood from 9–10 weeks onward; screens for trisomies 21, 18, 13, sex chromosome aneuploidies, and sometimes select microdeletions. Per ACMG cfDNA screening recommendations, cfDNA is the most sensitive and specific screening option for common aneuploidies and should be offered to all pregnant patients. cfDNA is a screen, not a diagnostic test — positive results require confirmation with CVS or amniocentesis.
Diagnostic Procedures
Chorionic Villus Sampling (CVS): transabdominal or transcervical sampling of placental tissue at 10–13 weeks. Procedure-related pregnancy loss ~0.1–0.2% in experienced centers. Used for karyotype, CMA, FISH, and targeted gene testing. Confined placental mosaicism is a known limitation.
Amniocentesis: transabdominal aspiration of amniotic fluid at ≥ 15 weeks. Same analytes plus AFP and acetylcholinesterase for neural tube defect assessment. Procedure-related loss ~0.1–0.3%.
Percutaneous Umbilical Blood Sampling (PUBS/cordocentesis): rarely used now; reserved for fetal anemia or specific hematologic questions.
Preimplantation Genetic Testing (PGT)
Performed on embryos created via IVF. PGT-A — aneuploidy screening of all 24 chromosomes. PGT-M — monogenic; targeted testing for a known familial variant (e.g., BRCA, Huntington, cystic fibrosis). PGT-SR — structural rearrangements (balanced translocation carriers).
Carrier Screening
Expanded pan-ethnic carrier screening is now standard. The ACMG 2021 carrier screening guideline recommends a single pan-ethnic panel tier 3 including all conditions with carrier frequency ≥ 1/200 (> 100 conditions). Historically required panels: CF, SMA, hemoglobinopathies, and fragile X in indicated populations.
Newborn Screening
Every U.S. state screens newborns for conditions from the federal Recommended Uniform Screening Panel (RUSP). Current RUSP includes 37+ core conditions: PKU, MSUD, homocystinuria, tyrosinemia, galactosemia, biotinidase deficiency, CAH, congenital hypothyroidism, MCAD and other FAODs, organic acidemias, sickle cell, CF, SCID, SMA, X-ALD, Pompe, MPS I, Krabbe (state-variable), and hearing loss. Positive screens trigger confirmatory testing and urgent genetics/metabolic evaluation.
18 Medications & Targeted Therapies Therapeutics
Genetic therapeutics have expanded dramatically in the last decade. Below are the drug classes a scribe will encounter in a modern genetics clinic.
| Drug Class | Example (Generic / Brand) | Indication | Mechanism |
|---|---|---|---|
| Enzyme replacement (ERT) | Imiglucerase (Cerezyme) | Gaucher type 1 | IV recombinant glucocerebrosidase |
| ERT | Agalsidase beta (Fabrazyme) | Fabry disease | IV recombinant alpha-galactosidase A |
| ERT | Laronidase (Aldurazyme) | MPS I | IV recombinant alpha-L-iduronidase |
| ERT | Idursulfase (Elaprase) | MPS II (Hunter) | IV recombinant iduronate-2-sulfatase |
| ERT | Alglucosidase alfa (Myozyme/Lumizyme) | Pompe | IV recombinant acid alpha-glucosidase |
| Substrate reduction (SRT) | Miglustat (Zavesca) | Gaucher, Niemann-Pick C | Inhibits glucosylceramide synthase |
| SRT | Eliglustat (Cerdelga) | Gaucher type 1 | Oral glucosylceramide synthase inhibitor |
| Pharmacological chaperone | Migalastat (Galafold) | Fabry (amenable variants) | Stabilizes mutant alpha-galactosidase A |
| Chaperone | Sapropterin (Kuvan) | PKU (BH4-responsive) | Cofactor for residual PAH activity |
| Enzyme substitution | Pegvaliase (Palynziq) | Adult PKU | PEGylated phenylalanine ammonia lyase |
| Nitrogen scavenger | Sodium phenylbutyrate, glycerol phenylbutyrate (Ravicti) | Urea cycle disorders | Alternate ammonia disposal |
| Antisense oligonucleotide | Nusinersen (Spinraza) | SMA | SMN2 splicing modification (intrathecal) |
| ASO | Eteplirsen, golodirsen, viltolarsen, casimersen | DMD (exon skipping) | Restores dystrophin reading frame |
| ASO | Inotersen (Tegsedi), tofersen (Qalsody) | hATTR amyloidosis; SOD1-ALS | Reduces target mRNA |
| siRNA | Patisiran (Onpattro), vutrisiran (Amvuttra) | hATTR amyloidosis | RNAi against TTR mRNA |
| TTR stabilizer | Tafamidis (Vyndaqel/Vyndamax) | ATTR cardiomyopathy/neuropathy | Stabilizes TTR tetramer |
| AAV gene therapy | Onasemnogene abeparvovec (Zolgensma) | SMA in infants | Single-dose SMN1 delivery via AAV9 |
| AAV gene therapy | Voretigene neparvovec (Luxturna) | RPE65 retinal dystrophy | Subretinal AAV delivery |
| AAV gene therapy | Delandistrogene moxeparvovec (Elevidys) | Duchenne | Micro-dystrophin delivery |
| Ex vivo gene therapy | Exa-cel (Casgevy), lovo-cel (Lyfgenia), beti-cel (Zynteglo) | Sickle cell, beta-thalassemia | CRISPR or lentiviral editing of autologous HSCs |
| CFTR modulator | Elexacaftor/tezacaftor/ivacaftor (Trikafta) | Cystic fibrosis | CFTR correctors + potentiator |
| Complement inhibitor | Eculizumab (Soliris), ravulizumab (Ultomiris) | PNH, aHUS | Anti-C5 monoclonal antibody |
| PARP inhibitor | Olaparib, talazoparib | BRCA-associated cancers | Synthetic lethality in HR-deficient tumors |
| mTOR inhibitor | Everolimus, sirolimus | TSC (SEGA, AML, seizures) | mTOR pathway inhibition |
| MEK inhibitor | Selumetinib (Koselugo) | NF1 plexiform neurofibromas | Inhibits RAS/MAPK pathway |
| HIF-2 inhibitor | Belzutifan (Welireg) | VHL-associated tumors | HIF-2alpha inhibition |
| CNP analog | Vosoritide (Voxzogo) | Achondroplasia | Promotes endochondral growth |
Dietary management remains the cornerstone of many metabolic disorders: phenylalanine restriction and medical formula in PKU, BCAA restriction in MSUD, protein restriction with essential amino acid formulas in urea cycle disorders, and galactose restriction in galactosemia.
19 ACMG Variant Classification & Reporting Reporting
Every genetic test result you document uses the ACMG/AMP 2015 variant interpretation standards. Variants are classified on a 5-tier scale based on population, computational, functional, segregation, de novo, and allelic evidence.
| Tier | Clinical Meaning | Documentation Implication |
|---|---|---|
| Pathogenic (P) | Variant clearly causes disease; sufficient evidence | Actionable — guides management and cascade testing |
| Likely Pathogenic (LP) | >90% certainty of disease causation | Treated clinically similar to pathogenic |
| Variant of Uncertain Significance (VUS) | Insufficient evidence to classify | Do NOT change management based on a VUS; do not order cascade testing |
| Likely Benign (LB) | >90% certainty of no disease causation | Generally not reported in clinical context |
| Benign (B) | Clearly not disease-causing | Not clinically actionable |
Evidence codes include PVS1 (null variant in loss-of-function gene), PS1–4 (strong evidence: same amino acid change, de novo confirmed, functional study, prevalence), PM1–6 (moderate evidence), PP1–5 (supporting evidence: segregation, missense in constrained gene, reputable source). Benign codes run BA1, BS1–4, BP1–7. Resources such as ClinVar and ClinGen expert panels publish curated variant assessments that supersede individual lab calls.
Never paraphrase a VUS as "positive" or "abnormal." A VUS is uncertain by definition. The clinician will often say, "We don't change management for a VUS. We'll re-review in 1–2 years as the evidence evolves." Document that language exactly.
ACMG Secondary Findings
When exome or genome sequencing is performed, the ACMG recommends reporting of medically actionable secondary findings in a defined gene list (currently ~78 genes in SF v3.2) covering hereditary cancer, cardiovascular, and metabolic conditions unless the patient opts out during consent.
20 Dysmorphology Physical Exam
The dysmorphology exam is the most specialty-specific physical exam in medicine. Clinical geneticists and dysmorphologists systematically describe every body region using standardized terminology from the Elements of Morphology consensus. Your job is to transcribe these terms accurately.
Growth: Height, weight, head circumference in cm and percentiles; proportionality assessment (sitting height, arm span, upper/lower segment ratio).
Head / Skull: Macrocephaly (> 97th), microcephaly (< 3rd), brachycephaly, dolichocephaly, plagiocephaly, frontal bossing, biparietal narrowing, open/closed fontanelles.
Face: Triangular, round, coarse, flat, or long facies. Midface hypoplasia or retrusion.
Eyes: Hypertelorism (increased interpupillary distance), hypotelorism, telecanthus, upslanting or downslanting palpebral fissures, epicanthal folds, ptosis, proptosis, synophrys, long/sparse eyelashes, blue sclerae, iris anomalies (Lisch nodules, Brushfield spots, coloboma), cataracts, lens dislocation.
Ears: Low-set (below line from outer canthus), posteriorly rotated, cupped, overfolded helix, preauricular pits/tags, microtia.
Nose: Broad or depressed nasal bridge, bulbous tip, anteverted nares, short columella, choanal atresia.
Mouth: Thin or full vermilion, long or short philtrum, smooth philtrum (FASD), micrognathia, retrognathia, macroglossia, high-arched or cleft palate, bifid uvula, natal teeth, gum hypertrophy.
Neck: Short, webbed, low posterior hairline, branchial cleft remnants.
Chest: Pectus excavatum or carinatum, widely spaced nipples, shield chest.
Abdomen: Hepatomegaly, splenomegaly, umbilical hernia, diastasis.
Genitourinary: Ambiguous genitalia, micropenis, hypospadias, cryptorchidism, clitoromegaly.
Extremities: Arachnodactyly (wrist/thumb signs), brachydactyly, clinodactyly (incurved fifth finger), camptodactyly (flexed finger), syndactyly, polydactyly, single transverse palmar crease, sandal gap toe, rocker-bottom feet, pes cavus/planus, joint hypermobility (Beighton score), contractures.
Skin: Cafe-au-lait macules (count, size, location), hypopigmented macules, hemangiomas, nevi, angiokeratomas, axillary/inguinal freckling, shagreen patch, ash-leaf macules, skin hyperextensibility, atrophic scars.
Neurologic: Tone (hypotonia/hypertonia), deep tendon reflexes, primitive reflexes in infants, cranial nerves, gait assessment, developmental milestones.
Beighton Hypermobility Score
| Maneuver | Points |
|---|---|
| Passive fifth MCP hyperextension > 90° | 1 per side (2 max) |
| Passive thumb apposition to forearm | 1 per side (2 max) |
| Elbow hyperextension > 10° | 1 per side (2 max) |
| Knee hyperextension > 10° | 1 per side (2 max) |
| Forward trunk flexion with palms flat on floor, knees extended | 1 |
| Total | /9 |
A score ≥ 5 in adults (≥ 6 prepubertal, ≥ 4 > 50 years) meets the hypermobility criterion used in the 2017 hEDS classification.
21 Abbreviations Master List
Inheritance & Molecular
Tests & Procedures
Conditions & Syndromes
Clinical & Regulatory
22 Sample HPI Templates
These HPIs reflect the pace and content of a real medical genetics clinic. Use them as scaffolds.
"Ms. [Name] is a 38-year-old G2P2 female of Ashkenazi Jewish ancestry referred for hereditary cancer risk assessment. She is personally unaffected. Family history is notable for: mother diagnosed with bilateral breast cancer at ages 44 and 51 (status post bilateral mastectomy, alive at 68); maternal aunt diagnosed with ovarian cancer at age 49 (deceased at 52); maternal grandmother diagnosed with breast cancer at age 60; maternal uncle diagnosed with pancreatic cancer at age 58. No known genetic testing has been performed in the family. The patient's personal cancer screening is up to date, including normal annual mammograms starting at age 30 and a normal pelvic exam last month. She meets NCCN criteria for hereditary breast and ovarian cancer testing based on Ashkenazi Jewish ancestry and multiple affected first/second-degree relatives with early-onset breast and ovarian cancer. Pre-test counseling was provided regarding BRCA1/2 and expanded hereditary cancer panel testing, possible results, GINA protections, and implications for first-degree relatives."
"[Name] is a 3-year-old male referred by his pediatrician for global developmental delay and dysmorphic features. He was born at 39 weeks via uncomplicated SVD to a 32-year-old G2P1 mother; birth weight 3.1 kg (25th%), length 49 cm, HC 34 cm. Prenatal course unremarkable; NIPT low risk. Hypotonia was noted in the newborn period. Developmental history: sat at 11 months, walked at 22 months, currently uses < 10 single words with no two-word phrases. Receives speech, OT, and PT through Early Intervention. No seizures. No regression. ROS notable for recurrent otitis media and constipation. Family history: nonconsanguineous parents of Northern European ancestry, no known intellectual disability, recurrent miscarriages, or congenital anomalies. Exam today: weight 14 kg (50th%), height 92 cm (25th%), HC 47 cm (3rd%). Dysmorphic features include epicanthal folds, upslanting palpebral fissures, flat nasal bridge, low-set posteriorly rotated ears, high-arched palate, bilateral fifth-finger clinodactyly, and generalized hypotonia. Prior testing: normal fragile X, normal CMA. Plan: proceed with trio exome sequencing after insurance preauthorization and pre-test counseling."
"Mrs. [Name] is a 34-year-old G3P2 female at 12+3 weeks by LMP (confirmed by 8-week dating ultrasound) referred for genetic counseling after a high-risk cell-free DNA result. NIPT (Natera Panorama) reported a T21 risk of > 99/100 with a fetal fraction of 9.6%. First-trimester ultrasound today demonstrates a single live intrauterine pregnancy with nuchal translucency of 3.8 mm, absent nasal bone, and otherwise normal fetal anatomy for gestational age. Prior obstetric history: two healthy term children, no prior pregnancy losses or anomalies. Family history: noncontributory, no consanguinity, no known genetic conditions. The limitations of cfDNA as a screening test and the implications of the high-risk result were discussed in detail, including the need for diagnostic confirmation via CVS or amniocentesis, procedure-related pregnancy loss risk, and alternative testing strategies. The patient elected to proceed with CVS today with karyotype, CMA, and FISH for rapid aneuploidy detection. Post-procedure options including continuation of pregnancy with planning for a child with Down syndrome versus termination were introduced as future discussions pending definitive results."
"[Name] is a 4-day-old term male (39+2 weeks, birth weight 3.4 kg) admitted to the NICU for encephalopathy and hyperammonemia. He was born via uncomplicated SVD to nonconsanguineous parents and discharged at 36 hours of life. At home, he fed well for the first 48 hours, then developed progressive lethargy, poor feeding, and one episode of emesis. On presentation to the ED, he was obtunded with tachypnea and a plasma ammonia of 612 µmol/L (reference < 50). Initial labs: normal anion gap (12), lactate 2.1, glucose 68, no ketones. Plasma amino acids and urine organic acids have been sent. He was started on protein restriction, IV dextrose, and nitrogen scavengers (sodium benzoate/sodium phenylacetate) with hemodialysis underway for ammonia clearance. Newborn screen pending. Family history: previously healthy older sister; maternal cousin died in infancy from 'a metabolic condition, details unknown.' Differential includes urea cycle disorder (OTC deficiency most likely given pattern), organic acidemia, and fatty acid oxidation defect. Plan: rapid exome sequencing through the NICU rapid pipeline and targeted OTC analysis pending biochemical results."
"Ms. [Name] is a 45-year-old female previously seen in this clinic for personal history of triple-negative breast cancer diagnosed at age 41, returning today to discuss results of a 76-gene hereditary cancer panel (Invitae). Results: a pathogenic variant in BRCA1 (c.68_69delAG, p.Glu23ValfsTer17), a known Ashkenazi Jewish founder variant. No additional pathogenic variants, one VUS in ATM (c.5557G>A, p.Asp1853Asn) noted. Results were reviewed in detail including the clinical significance of the BRCA1 variant, the 65–72% lifetime breast cancer risk, 39–44% ovarian cancer risk, and elevated risks for contralateral breast and pancreatic cancer. Risk-reducing strategies discussed: ongoing high-risk breast screening with annual MRI and mammogram, consideration of contralateral risk-reducing mastectomy, and recommendation for risk-reducing bilateral salpingo-oophorectomy by age 40–45 (patient is currently 45). The ATM VUS was explained as uncertain and not used to guide management. Cascade testing for the patient's two adult sisters, three adult daughters, and living parents was strongly recommended, and a family letter was provided. GINA protections and implications for life/disability insurance were reviewed."
"Mr. [Name] is a 28-year-old male referred for evaluation of possible connective tissue disorder after an echocardiogram performed for an atypical chest pain workup demonstrated an aortic root diameter of 4.3 cm (Z-score +3.1). He reports lifelong tall stature (6'5''), long limbs, pectus excavatum repaired at age 14, mild scoliosis, and chronic joint pain. He denies lens dislocation, prior spontaneous pneumothorax, or dural ectasia symptoms. Family history: father 6'4'' with known pectus excavatum and a 'murmur,' deceased at age 52 from sudden cardiac death (no autopsy); paternal uncle with aortic surgery at age 48; paternal grandmother tall and 'thin.' Exam notable for tall marfanoid habitus, arm span exceeding height by 7 cm, positive wrist and thumb signs, high-arched palate, pectus scar, mild scoliosis, and reduced upper/lower segment ratio. No striae, no hernias. Ophthalmology evaluation pending to assess for ectopia lentis. He meets revised Ghent criteria for Marfan syndrome based on aortic root Z-score ≥ 2 plus systemic score > 7. Plan: FBN1 sequencing with deletion/duplication analysis (or comprehensive thoracic aortic aneurysm panel to cover FBN1, TGFBR1/2, SMAD3, ACTA2, and related genes), initiation of losartan for aortic protection pending cardiology input, activity restriction counseling (no isometric or contact sports), and coordination with cardiothoracic surgery for serial aortic imaging."
23 References & Sources
Clinical Practice Guidelines
Gupta S, et al. NCCN Guidelines: Genetic/Familial High-Risk Assessment: Colorectal. JNCCN. 2022.
Allanson JE, et al. Elements of morphology: standard terminology. Am J Med Genet A. 2009.
ACMG Newborn Screening / RUSP updates, ACMG statements on newborn screening.
HRSA Recommended Uniform Screening Panel (RUSP).
ClinGen Resource. Expert variant and gene curation.
ClinVar. NCBI public archive of variant-phenotype relationships.
Diagram & Figure Sources
Figure 1: DNA double helix. Zephyris, Wikimedia Commons. CC BY-SA 3.0.
Figure 2: Normal human male karyotype. NHGRI, Wikimedia Commons. Public domain.
Figure 3: Autosomal dominant pedigree. U.S. NLM, Wikimedia Commons. Public domain.
Figure 4: Autosomal recessive pedigree. U.S. NLM, Wikimedia Commons. Public domain.
Figure 5: X-linked recessive pedigree. U.S. NLM, Wikimedia Commons. Public domain.
Figure 6: Pedigree chart example. Wikimedia Commons. Public domain.
Figure 7: Down syndrome karyotype (47,XY,+21). NHGRI, Wikimedia Commons. Public domain.
Medical genetics is the most vocabulary-dense specialty you will encounter. Gene names, variant nomenclature, syndrome eponyms, enzyme deficiencies, and inheritance patterns come in rapid succession. The great scribe learns to hear "c.68_69delAG in BRCA1" and chart it without asking. They recognize that "likely pathogenic" and "pathogenic" are managed similarly, while a "VUS" must never be conflated with either. They know that a family history HPI is only as good as the ages at diagnosis and causes of death included.
Use this guide as a starting map. Learn the syndromes that come through your clinic most often first, then work outward. Keep a personal cheat sheet of the labs your clinic uses, the specific panel names, and the referring providers. Read ClinVar, GeneReviews, and OMIM entries for conditions you see repeatedly — the more you understand the biology, the better your notes become.
Genetics is uniquely high-stakes because the results do not just affect one patient — they affect entire families across generations. Accurate documentation is not a clerical task here. It is a piece of durable medical knowledge that cascade testing, surveillance, and reproductive decisions will rely on for decades. Welcome to medical genetics.