Pediatric Surgery
Every diagnosis, classification, procedure, technique, medication, complication, and management algorithm across the full scope of pediatric surgery in one place.
01 Pediatric Surgical Anatomy
Children are not small adults. The pediatric surgical patient has unique anatomical features at every stage of development, from the premature neonate through the adolescent. Understanding these differences is essential for safe operative planning.
Airway
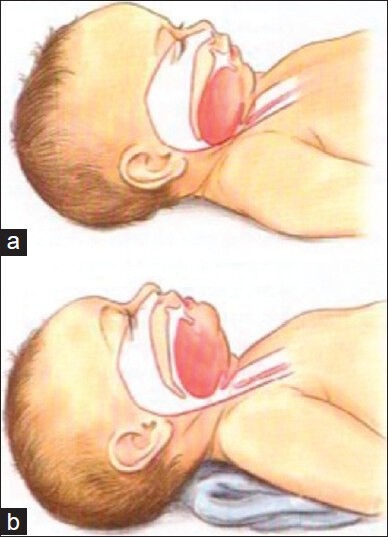
The neonatal airway is markedly different from the adult. The larynx is higher (C3-C4 vs C4-C6 in adults), the epiglottis is omega-shaped and floppy, and the narrowest point is the cricoid ring (subglottic — unlike adults where the glottis is narrowest). The trachea is short (4-5 cm in neonates vs 11-13 cm in adults), making right mainstem intubation and accidental extubation common. Uncuffed ETTs are traditionally used in children < 8 years (the circular cricoid provides a natural seal), though modern low-pressure cuffed tubes are increasingly used. ETT size formula: (age/4) + 4 for uncuffed; (age/4) + 3.5 for cuffed.
Chest Wall & Lungs
The infant chest wall is highly compliant (cartilaginous ribs) with horizontal ribs, making intercostal muscle contribution to breathing minimal. Neonates are obligate diaphragmatic breathers — anything that impedes diaphragmatic excursion (abdominal distension, diaphragmatic hernia) causes rapid respiratory compromise. Functional residual capacity (FRC) is low relative to oxygen consumption, so desaturation occurs rapidly during apnea (safe apnea time ~90 seconds in a neonate vs 5-8 minutes in an adult).
Abdominal Wall
The neonatal abdominal wall is thin with poorly developed musculature. The inguinal canal is short (~1 cm in neonates vs 4 cm in adults), and the internal and external rings nearly overlie each other. The umbilical ring closes by age 5 in most children. The processus vaginalis (a peritoneal outpouching accompanying testicular descent) remains patent in up to 80% of neonates, closing spontaneously in most — persistent patency leads to indirect inguinal hernia or communicating hydrocele.
Gastrointestinal Tract
The neonatal stomach holds only 10-20 mL; the lower esophageal sphincter is immature, predisposing to gastroesophageal reflux. The midgut undergoes a complex 270-degree counterclockwise rotation during weeks 4-12 of gestation around the SMA axis — failure of this process produces malrotation. The appendix is relatively longer and more mobile in children, and cecal position may be higher than in adults. Intestinal length: approximately 250 cm at birth (term), growing to 600-800 cm in adults.
Hepatobiliary
The neonatal liver is relatively large (4% of body weight vs 2% in adults), extending below the costal margin. Hepatic enzyme systems are immature — drug metabolism (especially glucuronidation) is reduced, and bilirubin conjugation is inefficient (physiologic jaundice). The extrahepatic biliary tree is small and delicate — the common bile duct is only 1-2 mm in a neonate, making operative manipulation technically demanding.
Genitourinary
The kidneys are lobulated at birth and relatively large. The bladder is an abdominal organ in infants (lies above the pubic symphysis), only becoming a true pelvic organ by age 6 — important for suprapubic catheter placement (easier in infants) and during laparotomy (avoid bladder injury). Renal function matures over the first 1-2 years: neonatal GFR is approximately 20 mL/min/1.73 m2 at birth, reaching adult levels (~120 mL/min/1.73 m2) by age 2. Concentrating ability is limited — maximum urine osmolality is ~600 mOsm/kg in neonates vs 1200 mOsm/kg in adults, making neonates susceptible to dehydration and less able to handle fluid overload. Normal urine output: neonates 1-3 mL/kg/hr; infants/children 1-2 mL/kg/hr.
02 Pediatric Physiology & Fluid Management
Fluid Requirements — The 4-2-1 Rule (Holliday-Segar)
Maintenance intravenous fluid calculation for children by weight:
| Body Weight | Rate | Example |
|---|---|---|
| First 10 kg | 4 mL/kg/hr | 10 kg child = 40 mL/hr |
| Next 10 kg (11-20 kg) | 2 mL/kg/hr | 20 kg child = 40 + 20 = 60 mL/hr |
| Each additional kg (>20 kg) | 1 mL/kg/hr | 30 kg child = 40 + 20 + 10 = 70 mL/hr |
Standard maintenance fluid: D5 0.45% NS + 20 mEq/L KCl for children >1 month. Neonates (<1 month): D10W initially, transitioning to D5 0.2% NS. Isotonic fluids (NS or LR) are used for bolus resuscitation: 20 mL/kg bolus, repeated up to 3 times (60 mL/kg total) before considering blood products or vasopressors.
Electrolyte Differences
Neonates have higher total body water (~80% vs 60% in adults) and a larger extracellular fluid compartment. Sodium: maintenance requirement is 2-3 mEq/kg/day. Potassium: 1-2 mEq/kg/day. Premature infants have immature renal tubular function with obligate sodium wasting — may require sodium supplementation. Hyponatremia is the most common electrolyte abnormality in hospitalized children.
Thermoregulation
Neonates have a high surface area-to-body mass ratio, thin skin, minimal subcutaneous fat, and limited ability to shiver. Heat loss occurs primarily through radiation (most significant), convection, conduction, and evaporation. Hypothermia causes increased oxygen consumption, metabolic acidosis, apnea, and coagulopathy. The OR must be warmed to 26-28°C for neonatal surgery, with warming devices (forced-air warmers, warming mattress, warmed irrigation) used throughout.
Hypothermia in neonates increases morbidity and mortality significantly. For every 1°C drop in core temperature, oxygen consumption rises by 10%. A cold neonate develops metabolic acidosis, hypoglycemia, pulmonary vasoconstriction, and coagulopathy. Prevention is paramount: raise OR temperature, use radiant warmers, warm all fluids, minimize exposed body surface area, and use plastic wraps for premature infants.
Glucose Homeostasis
Neonates have limited glycogen stores and high glucose utilization rates (6-8 mg/kg/min). Hypoglycemia (<40 mg/dL in neonates) must be prevented and aggressively treated — it causes seizures, brain injury, and death. Treatment of acute hypoglycemia: D10W 2-4 mL/kg IV bolus, followed by continuous glucose infusion (GIR — glucose infusion rate) at 6-8 mg/kg/min. Monitor glucose every 30-60 minutes until stable. All neonates undergoing surgery require glucose-containing IV fluids and serial glucose monitoring (every 1-2 hours intraoperatively). Populations at highest risk: SGA (small-for-gestational-age), LGA (large-for-gestational-age), premature infants, infants of diabetic mothers, and infants with Beckwith-Wiedemann syndrome.
Hyperglycemia (>250 mg/dL) can also occur in stressed neonates, particularly premature infants on TPN with high dextrose concentrations. Risks include osmotic diuresis, dehydration, and increased risk of intraventricular hemorrhage. Treat by reducing the GIR; insulin infusion (0.01-0.1 units/kg/hr) is reserved for refractory cases.
Hematologic Considerations
Blood volume varies by age: premature infants ~100 mL/kg, term neonates ~80 mL/kg, infants ~75 mL/kg, children ~70 mL/kg. A 3 kg neonate has only ~240 mL of total blood volume — small losses are proportionally significant. Hgb at birth is 14-20 g/dL (fetal hemoglobin predominates), with a physiologic nadir at 8-12 weeks (Hgb ~10 g/dL). Transfusion threshold: typically Hgb <7 g/dL for stable patients, <10 g/dL for neonates with cardiopulmonary disease. PRBCs are transfused at 10-15 mL/kg.
03 Neonatal Assessment & Prematurity
Apgar Score
| Parameter | 0 | 1 | 2 |
|---|---|---|---|
| Appearance (color) | Blue/pale all over | Acrocyanosis (blue extremities) | Completely pink |
| Pulse (heart rate) | Absent | <100 bpm | ≥100 bpm |
| Grimace (reflex) | No response | Grimace | Cry/cough/sneeze |
| Activity (muscle tone) | Limp | Some flexion | Active motion |
| Respiration | Absent | Slow/irregular | Good cry |
Scored at 1 and 5 minutes of life. Score 7-10 = reassuring; 4-6 = moderately depressed; 0-3 = severely depressed. The 5-minute score correlates better with neonatal outcomes. Apgar does not guide resuscitation — NRP guidelines do.
Gestational Age & Prematurity
| Classification | Gestational Age | Key Surgical Implications |
|---|---|---|
| Extremely preterm | <28 weeks | Highest NEC risk, IVH, RDS, PDA, ROP; skin fragile, minimal reserves |
| Very preterm | 28-31 weeks | Significant NEC risk, BPD, need for NICU; inguinal hernia common |
| Moderate preterm | 32-33 weeks | Apnea of prematurity, feeding difficulties, moderate NEC risk |
| Late preterm | 34-36 weeks | Respiratory issues, hypoglycemia; often near-term appearance is deceptive |
| Term | 37-41 weeks | Full physiologic maturity expected by 39 weeks |
Prematurity-Related Surgical Conditions
Necrotizing enterocolitis (NEC) — the most common surgical emergency in premature infants. Patent ductus arteriosus (PDA) — may require ligation if medical therapy (indomethacin, ibuprofen) fails. Inguinal hernia — incidence of 9-11% in premature infants (vs 1-5% in term), with high incarceration risk. Retinopathy of prematurity (ROP) — laser therapy or anti-VEGF treatment. Intraventricular hemorrhage (IVH) — may require ventriculoperitoneal shunt placement.
04 Pediatric Surgical Principles
Several principles distinguish pediatric from adult surgical practice and must guide every operative decision.
Minimally Invasive Surgery
Laparoscopy and thoracoscopy are increasingly used in children. Smaller instruments (3 mm, 5 mm ports), lower insufflation pressures (8-12 mmHg in infants vs 15 mmHg in adults), and shorter working distances require adapted techniques. Advantages include less postoperative pain, faster recovery, improved cosmesis, and fewer adhesions. Common pediatric laparoscopic procedures: appendectomy, cholecystectomy, pyloromyotomy, fundoplication, inguinal hernia repair, splenectomy, and pull-through for Hirschsprung disease.
Tissue Handling
Pediatric tissues are delicate and more prone to injury from aggressive handling. Fine instruments and minimal tissue manipulation are essential. Electrocautery must be used judiciously — thermal spread in small structures can damage adjacent organs. Absorbable sutures are preferred for growing tissues. Nonabsorbable materials (mesh, permanent sutures) are generally avoided in young children due to growth concerns and foreign body reaction.
Growth Considerations
Every pediatric surgical procedure must account for future growth. Rigid repairs (tight closures, nonabsorbable sutures at the abdominal wall) may cause obstruction as the child grows. Bowel anastomoses must be generous — a caliber mismatch between proximal dilated and distal unused bowel is common in neonatal surgery; techniques such as fish-mouth or back-cut on the smaller end improve the anastomosis. Esophageal replacements must have growth potential. Vascular repairs in infants may require revision as the child outgrows the original reconstruction. Mesh should be avoided in young children when possible (does not expand with growth; can erode or become infected).
Nutritional Support
Caloric requirements are high: neonates require 100-120 kcal/kg/day, infants 90-100 kcal/kg/day. Enteral feeding is always preferred over parenteral nutrition when feasible — it preserves gut mucosal integrity, reduces infection risk, and prevents TPN-associated liver disease. TPN-associated cholestasis is a significant concern in neonates on prolonged parenteral nutrition (onset typically 2-4 weeks), managed by cycling TPN, reducing lipid infusion, and initiating even trickle enteral feeds as soon as possible.
Key questions before every pediatric surgical procedure: (1) Can this wait? Many conditions resolve spontaneously (umbilical hernia, physiologic phimosis). (2) Will the child grow out of it? (3) Is the anatomy mature enough for definitive repair? (4) Are the physiologic reserves adequate to tolerate the operation? (5) Will the repair accommodate growth?
05 Esophageal Atresia & Tracheoesophageal Fistula
Esophageal atresia (EA) occurs in approximately 1 in 3,000-4,500 live births. The esophagus ends in a blind pouch, with or without a fistulous connection to the trachea (tracheoesophageal fistula, TEF).
Gross Classification
| Type | Description | Frequency |
|---|---|---|
| Type A | Pure EA, no fistula (long gap) | 8-10% |
| Type B | EA with proximal TEF | 1-2% |
| Type C | EA with distal TEF | 85% (most common) |
| Type D | EA with both proximal and distal TEF | 1-2% |
| Type E (H-type) | TEF without EA (isolated fistula) | 4-5% |
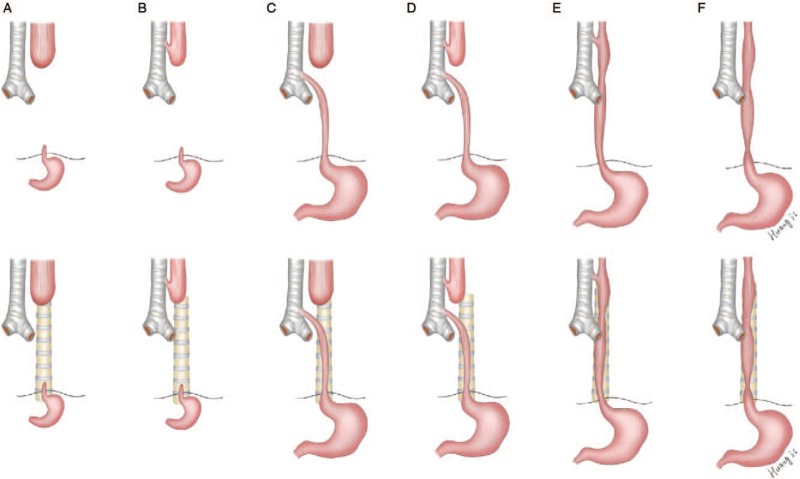
Clinical Presentation
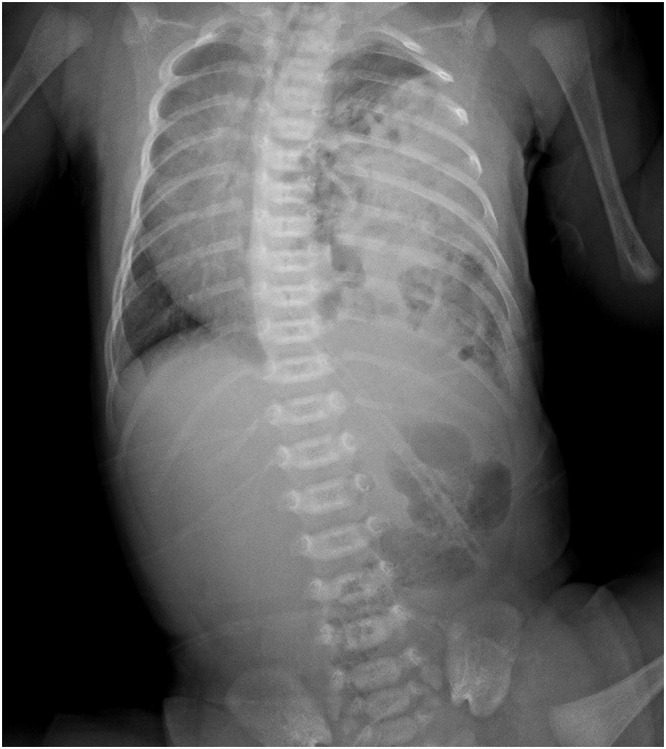
Prenatal: polyhydramnios (inability to swallow amniotic fluid — especially Type A). Postnatal: excessive drooling, choking, cyanosis with first feeding attempt, inability to pass a nasogastric tube (it coils in the blind pouch — typically at 10-12 cm from the nares). On chest X-ray: the NG tube coils in the upper pouch. Gas in the stomach/bowel indicates a distal fistula (Type C). Gasless abdomen suggests no distal fistula (Type A).
VACTERL Association
Up to 50% of EA/TEF patients have associated anomalies. VACTERL: Vertebral (hemivertebrae), Anorectal (imperforate anus), Cardiac (VSD, ASD, tetralogy of Fallot — most common and most lethal associated anomaly), TracheoEsophageal fistula, Renal (horseshoe kidney, renal agenesis), Limb (radial ray defects, thumb anomalies). Workup: echocardiogram, renal ultrasound, spinal X-ray, limb examination.
Spitz Classification (Prognostic)
| Group | Criteria | Survival |
|---|---|---|
| I | Birth weight >1500 g, no major cardiac defect | >97% |
| II | Birth weight <1500 g or major cardiac defect | ~59% |
| III | Birth weight <1500 g and major cardiac defect | ~22% |
Surgical Management
Type C (most common): Right extrapleural thoracotomy (or thoracoscopic approach) through the 4th intercostal space. The azygos vein is divided. The distal TEF is identified, ligated, and divided. The proximal esophageal pouch is mobilized, and a primary end-to-end esophageal anastomosis is performed. A transanastomotic feeding tube is placed.
Long-gap EA (Type A): Primary anastomosis is not possible. Options include: (1) delayed primary repair (Foker technique — apply traction sutures to stretch pouches over weeks), (2) esophageal replacement with gastric pull-up, colon interposition, or jejunal interposition. A gastrostomy tube is placed for feeding.
Complications
Anastomotic leak (5-15%) — often managed conservatively with drainage. Anastomotic stricture (30-40%) — the most common long-term complication, treated with serial esophageal dilations. Recurrent TEF (5-10%). Gastroesophageal reflux (40-70%) — nearly universal after repair, often requiring fundoplication. Tracheomalacia (10-20%) — from intrinsic tracheal cartilage deficiency at the fistula site; may cause life-threatening "dying spells" (reflex apnea with feeding) requiring aortopexy.
06 Congenital Diaphragmatic Hernia
Congenital diaphragmatic hernia (CDH) involves herniation of abdominal viscera into the chest through a diaphragmatic defect, causing pulmonary hypoplasia and pulmonary hypertension. Incidence: 1 in 2,500-5,000 live births. Mortality: 20-50% depending on severity.
Types
| Type | Location | Frequency | Features |
|---|---|---|---|
| Bochdalek | Posterolateral | 80-90% | Left-sided 80%, right-sided 20%; most common CDH |
| Morgagni | Anterior (retrosternal) | 5-10% | Right-sided predominance; often asymptomatic, incidental finding |
| Eventration | Thinned diaphragm | Rare | Diaphragm present but attenuated; may mimic CDH |
Pathophysiology
The key pathology is pulmonary hypoplasia (both ipsilateral and contralateral lungs are underdeveloped) and persistent pulmonary hypertension of the newborn (PPHN). The hypoplastic lungs have fewer alveoli and pulmonary arterioles with abnormal muscularization, leading to increased pulmonary vascular resistance. This is not merely a "hole in the diaphragm" — it is a disease of lung development.
Clinical Presentation
Most diagnosed prenatally on ultrasound (stomach/bowel in chest, mediastinal shift). Postnatal: respiratory distress at birth, scaphoid abdomen (viscera in chest), barrel-shaped chest, absent breath sounds on affected side, heart sounds shifted to contralateral side. CXR: bowel gas pattern in the chest, mediastinal shift.
Bag-mask ventilation forces air into the stomach and herniated bowel, further compressing the lungs. Immediate intubation is required. Place an OG/NG tube to decompress the stomach. Ventilate with gentle settings (permissive hypercapnia strategy — pH >7.25, PaCO2 <65 mmHg) to avoid barotrauma to the hypoplastic lungs.
Prognostic Indicators
Prenatal: lung-to-head ratio (LHR) — observed-to-expected LHR (O/E LHR) <25% = severe (mortality >80%), 25-45% = moderate, >45% = mild. Liver herniation into the chest worsens prognosis. CDH Study Group uses defect size (A-D) to predict outcomes.
Management
Stabilize first, operate later. CDH is no longer considered an immediate surgical emergency. Initial management focuses on cardiopulmonary stabilization: gentle ventilation (HFOV if conventional ventilation fails), inhaled nitric oxide for PPHN, sedation, inotropic support. ECMO criteria: failure to maintain preductal SpO2 >85% or PaO2 >60 mmHg despite maximal medical therapy, oxygenation index (OI) >40, progressive hemodynamic instability. ECMO is used as a bridge to stabilization, with repair performed either on or after ECMO decannulation.
Surgical repair: Subcostal abdominal approach (or thoracoscopic in stable patients). Abdominal viscera are carefully reduced from the chest into the abdomen. The diaphragmatic defect is assessed: small-to-moderate defects (A-B) are closed primarily with nonabsorbable interrupted horizontal mattress sutures. Large defects (C-D) lacking a posterior rim require a prosthetic patch (Gore-Tex or biological mesh such as Surgisis/AlloDerm). Patch repairs carry a higher recurrence rate (up to 40% for type D defects). Chest tubes are not routinely placed on the ipsilateral side (risk of creating negative pressure that causes mediastinal shift before the hypoplastic lung can expand). A contralateral chest tube may be placed for pneumothorax monitoring. The abdomen may be difficult to close if the viscera were chronically in the chest (underdeveloped abdominal cavity) — a silo or temporary closure may be necessary.
Outcomes: Overall survival 60-80% in high-volume centers. Major morbidity includes chronic lung disease (50%), pulmonary hypertension requiring long-term sildenafil, GERD (40-80% — may require fundoplication), neurodevelopmental delay, musculoskeletal deformities (scoliosis, pectus), hearing loss, and hernia recurrence. Long-term multidisciplinary follow-up is essential.
07 Intestinal Atresia
Duodenal Atresia
Incidence: 1 in 5,000-10,000 live births. 30% associated with Down syndrome (trisomy 21). Other associations: annular pancreas, malrotation, cardiac defects. Embryology: failure of recanalization of the duodenal lumen (weeks 8-10).
Classic finding: "Double bubble" sign on abdominal X-ray — the first bubble is the stomach, the second is the dilated proximal duodenum. No distal gas (complete obstruction) or minimal distal gas (duodenal web/stenosis). Prenatal ultrasound may show polyhydramnios and double bubble.
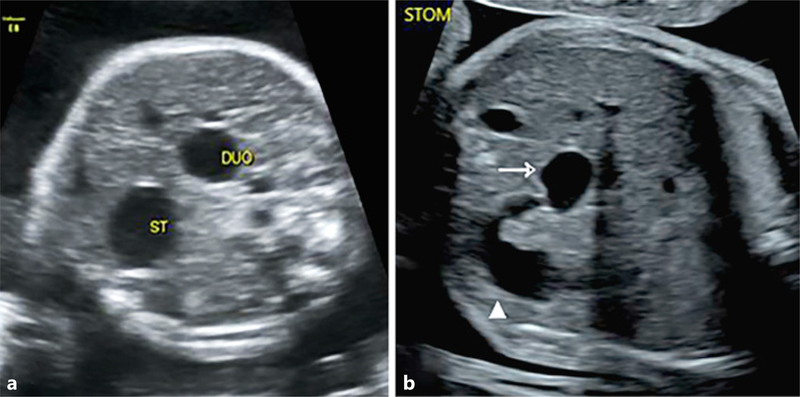
Treatment: Duodenoduodenostomy (diamond-shaped anastomosis — proximal transverse to distal longitudinal incision). The annular pancreas, if present, is not divided (risk of pancreatic fistula and injury to the pancreatic duct). A transanastomotic feeding tube is placed. Outcome is excellent (>95% survival).
Jejunoileal Atresia
Incidence: 1 in 5,000 live births. Etiology: intrauterine vascular accident (mesenteric vascular disruption) — unlike duodenal atresia, which is a failure of recanalization. No association with chromosomal anomalies.
| Type | Description | Frequency |
|---|---|---|
| Type I | Mucosal web/membrane, intact mesentery | 20% |
| Type II | Blind ends connected by fibrous cord, intact mesentery | 35% |
| Type IIIa | Blind ends separated by V-shaped mesenteric gap | 35% |
| Type IIIb | "Apple-peel" or "Christmas tree" — proximal jejunal atresia with absent SMA, distal bowel wraps around a marginal artery | 5% |
| Type IV | Multiple atresias ("string of sausages") | 5% |
Treatment: Resection of the dilated proximal segment (dysfunctional) with primary anastomosis. Tapering enteroplasty of the dilated segment may be performed to improve motility. In Type IIIb (apple-peel), bowel length is significantly reduced — short bowel syndrome is a major concern. Contrast enema preoperatively demonstrates a microcolon (unused distal bowel).
Colonic Atresia
Rarest form of intestinal atresia (<5% of all intestinal atresias). Also caused by intrauterine vascular accident. May be associated with gastroschisis or Hirschsprung disease. Treatment: resection with primary anastomosis or colostomy with delayed anastomosis.
08 Malrotation & Midgut Volvulus
Bilious (green) emesis in a neonate is malrotation with midgut volvulus until proven otherwise. This is a true surgical emergency — delay in diagnosis leads to midgut necrosis, short bowel syndrome, and death. An emergent upper GI series is the diagnostic study of choice. If high clinical suspicion, proceed directly to the operating room.
Normal Intestinal Rotation
During weeks 4-12 of gestation, the midgut herniates out of the abdominal cavity through the umbilical ring, undergoes a 270-degree counterclockwise rotation around the SMA, and returns to the abdomen. The duodenojejunal junction (ligament of Treitz) fixes in the left upper quadrant, and the cecum fixes in the right lower quadrant. The mesenteric base is broad (from ligament of Treitz to ileocecal valve), preventing volvulus.
Malrotation Pathology
When rotation is incomplete, the cecum remains in the right upper quadrant or midabdomen, and the duodenojejunal junction is displaced to the right. Ladd bands — peritoneal bands from the malpositioned cecum crossing the duodenum to the right upper quadrant — cause duodenal obstruction. The mesenteric base is narrow (the SMA pedicle), allowing the entire midgut to twist around this narrow axis (midgut volvulus). Volvulus produces obstruction of the duodenum and compromises the SMA blood supply to the entire midgut (from duodenum to transverse colon).
Diagnosis
Upper GI series is the gold standard — shows the duodenojejunal junction displaced to the right of the spine (normally should cross to the left at the ligament of Treitz at the level of the duodenal bulb). In volvulus: "corkscrew" or "bird's beak" pattern of the distal duodenum/proximal jejunum. Ultrasound may show inversion of SMA/SMV relationship (SMV to the left of or anterior to SMA — though position alone is unreliable; the "whirlpool sign" of twisted mesentery is more specific).
The Ladd Procedure
The definitive operation for malrotation, described by William Ladd in 1936:
- Eviscerate the bowel — deliver the entire midgut out of the abdomen and inspect for viability
- Detorse the volvulus — rotate the bowel counterclockwise (turning pages of a book) to untwist
- Divide Ladd bands — lyse the peritoneal bands crossing the duodenum from the cecum/right colon
- Widen the mesenteric base — separate the duodenum and colon, straightening the duodenum along the right gutter
- Appendectomy — performed because the cecum will be in an atypical position, making future appendicitis diagnosis confusing
- Place the bowel — small bowel on the right, colon on the left (nonrotation position)
If bowel viability is questionable: Warm the bowel, wait 15-20 minutes, and reassess. If extensive necrosis is present, resect only clearly necrotic bowel, leave questionable bowel, and perform a "second look" laparotomy in 24-48 hours to reassess viability and avoid unnecessary loss of bowel length.
09 Meconium Ileus & Abdominal Wall Defects
Meconium Ileus
Meconium ileus is intestinal obstruction caused by inspissated (abnormally thick and sticky) meconium in the terminal ileum. Almost pathognomonic for cystic fibrosis (CF) — 80-90% of neonates with meconium ileus have CF, and 15-20% of CF patients present with meconium ileus at birth.
Simple meconium ileus: Distal ileal obstruction, dilated proximal loops, microcolon. Treatment: Gastrografin enema (hyperosmolar water-soluble contrast) — the hyperosmolarity draws fluid into the bowel lumen, loosening the meconium. Successful in 60-80% of cases. Ensure adequate IV hydration before and during the procedure (risk of hypovolemia from fluid shifts).
Complicated meconium ileus: Volvulus, atresia, perforation, meconium peritonitis (meconium leaks into the peritoneal cavity causing intense inflammatory reaction; calcifications visible on abdominal X-ray). Requires surgical exploration: enterotomy with irrigation of inspissated meconium using warm saline or dilute N-acetylcysteine (Mucomyst), resection of atretic or necrotic segments, and temporary ileostomy. Stoma options include: Bishop-Koop (distal end brought out as stoma with proximal end-to-side anastomosis — allows antegrade flow while maintaining distal access for irrigation) or Santulli (proximal end brought out as stoma with distal end-to-side anastomosis). Primary anastomosis with T-tube chimney vent is an alternative. All infants with meconium ileus should undergo sweat chloride testing for CF diagnosis (typically performed at 2-4 weeks of age).
Gastroschisis
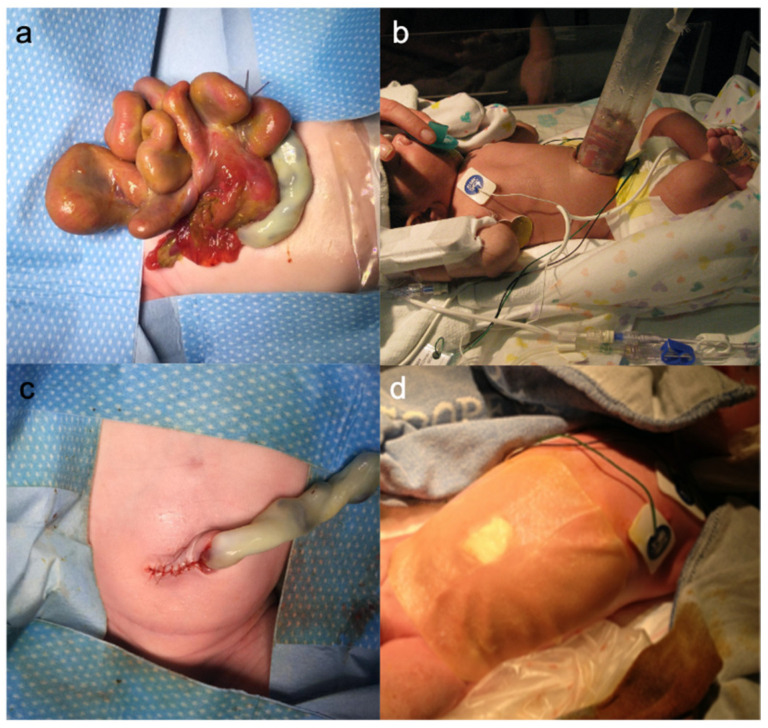
Full-thickness abdominal wall defect, typically right of the umbilicus, with evisceration of bowel (no covering membrane). Incidence: 1 in 4,000 births, increasing. Associations: young maternal age, smoking, vasoconstrictor use; notably not associated with chromosomal anomalies (unlike omphalocele).
The exposed bowel is edematous, matted, and covered in a fibrous inflammatory peel from prolonged exposure to amniotic fluid. Treatment: At birth, wrap bowel in warm saline-soaked gauze and place the lower body in a bowel bag to prevent heat and fluid loss. Decompress the stomach with an NG tube. Primary closure is ideal if the bowel can be reduced without causing abdominal compartment syndrome (monitor intragastric/intravesical pressure, ventilatory pressures). If reduction is not possible, a preformed silo (spring-loaded silo) is placed at bedside, and the bowel is gradually reduced over 3-7 days, followed by fascial closure.
Omphalocele
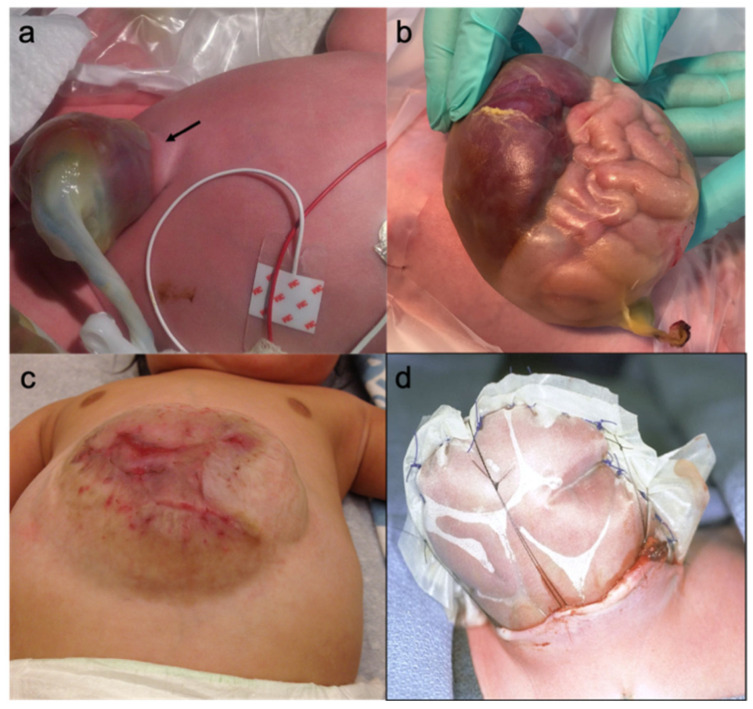
Midline abdominal wall defect at the umbilicus, with herniation of viscera covered by a sac (amnion-peritoneum membrane). Size ranges from small (containing only bowel loops) to giant (>5 cm, containing liver). Incidence: 1 in 5,000-10,000 births. High association with chromosomal anomalies (trisomy 13, 18, 21) and syndromes (Beckwith-Wiedemann — omphalocele, macroglossia, macrosomia, hypoglycemia, embryonal tumors). Associated cardiac anomalies in 30-50%.
Treatment: Small omphalocele: primary closure. Giant omphalocele: staged repair — paint the sac with silver sulfadiazine or antibiotic ointment to promote epithelialization (allowing a ventral hernia to form), followed by delayed definitive closure months to years later. Alternative: silo with serial reduction, then closure.
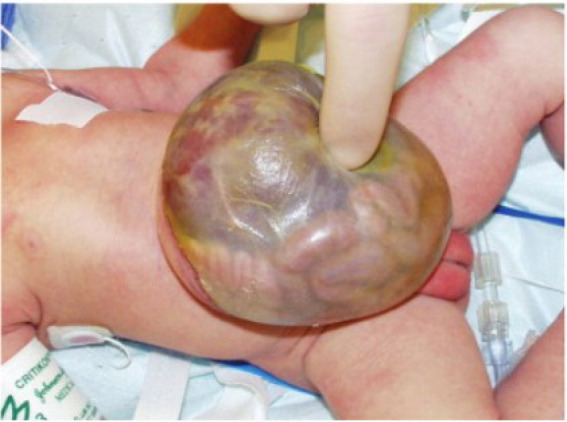
| Feature | Gastroschisis | Omphalocele |
|---|---|---|
| Location | Right of umbilicus | At the umbilicus |
| Sac | No sac (exposed bowel) | Covered by sac (membrane) |
| Contents | Bowel only (rarely liver) | Bowel, liver, spleen possible |
| Associated anomalies | Rare (intestinal atresia 10-15%) | Common (50-70%): chromosomal, cardiac |
| Maternal age | Young mothers (<20 y) | Older mothers (>35 y) |
| Prognosis | Good (>90% survival) | Depends on associated anomalies |
10 Anorectal Malformations & Hirschsprung Disease
Anorectal Malformations (ARM)
Spectrum of congenital anomalies affecting the anus and rectum, ranging from simple perineal fistula to complex cloacal malformations. Incidence: 1 in 4,000-5,000 live births. Males and females are equally affected, but the types differ.
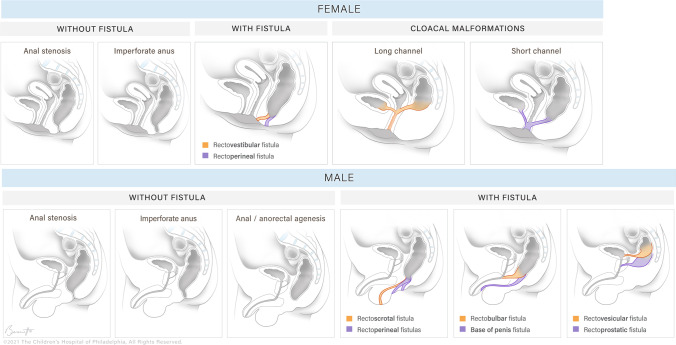
Krickenbeck Classification (2005)
| Males | Females |
|---|---|
| Perineal fistula (low) | Perineal fistula (low) |
| Rectourethral bulbar fistula | Vestibular fistula (most common in females) |
| Rectourethral prostatic fistula | Cloaca (common channel for urethra, vagina, rectum) |
| Rectobladder neck fistula (high) | Imperforate anus without fistula |
| Imperforate anus without fistula | Rectal atresia/stenosis |
| Rectal atresia/stenosis |
Initial management: (1) Examine the perineum — look for a fistula opening, meconium on the perineum, a "bucket-handle" deformity, or flat bottom (absence of gluteal cleft = poor prognosis for continence). (2) Obtain cross-table lateral prone X-ray (invertogram) or perineal ultrasound at 24 hours of life (allow time for air to reach the distal rectum). (3) Check for associated anomalies — VACTERL association is common. Echocardiogram, renal ultrasound, spinal ultrasound, sacral X-ray.
Surgical approach: Low lesions (perineal fistula): single-stage anoplasty in the neonatal period, no colostomy needed. Intermediate/high lesions: initial diverting colostomy (divided sigmoid colostomy), followed by definitive repair at 3-6 months of age via the posterior sagittal anorectoplasty (PSARP — Pena procedure). The PSARP involves a midline posterior sagittal incision, splitting the muscle complex precisely in the midline, identifying and mobilizing the rectum, and placing it within the center of the muscle complex and sphincter mechanism. Colostomy closure follows 2-3 months after PSARP.
Hirschsprung Disease
Congenital absence of ganglion cells (aganglionosis) in the distal bowel, beginning at the internal anal sphincter and extending proximally for a variable distance. Incidence: 1 in 5,000 live births. M:F = 4:1. The aganglionic segment fails to relax, creating a functional obstruction. The proximal normally ganglionated bowel dilates.
Extent of Disease
| Type | Extent | Frequency |
|---|---|---|
| Short-segment | Rectosigmoid (at or below the sigmoid) | 75-80% |
| Long-segment | Beyond the sigmoid colon | 15-20% |
| Total colonic | Entire colon (may extend into small bowel) | 5-8% |
Clinical Presentation
Neonatal: failure to pass meconium within 48 hours of birth (90% of term neonates pass meconium within 24 hours), abdominal distension, bilious emesis, refusal to feed. "Squirt sign" — explosive release of stool and gas on digital rectal examination (transient relief of obstruction). Older children: chronic constipation unresponsive to medical therapy, failure to thrive. Hirschsprung-associated enterocolitis (HAEC) is the most feared complication — fever, explosive diarrhea, abdominal distension, sepsis; can be lethal. Treated with rectal irrigations, broad-spectrum antibiotics, and decompression.
Diagnosis
Rectal suction biopsy is the gold standard — performed at bedside without anesthesia. The biopsy must be obtained at least 2-3 cm above the dentate line (below this level, ganglion cells are physiologically absent — the "hypoganglionic zone" — leading to false-positive results). Adequate tissue must include submucosa. Histologic findings: absence of ganglion cells in the submucosal (Meissner) and myenteric (Auerbach) plexuses, and hypertrophied nerve trunks (>40 microns). Acetylcholinesterase (AChE) staining shows increased activity in the lamina propria and muscularis mucosa. Calretinin immunostaining — absent in aganglionic tissue (normally present in ganglion cells and nerve fibers) — is increasingly used as a reliable adjunct. Contrast enema: may show a transition zone (narrow distal aganglionic segment to dilated proximal ganglionated segment) with caliber change and rectosigmoid ratio <1 (normally the rectum is wider than the sigmoid). Delayed 24-hour film showing retained contrast is suggestive. Note: contrast enema can be unreliable in neonates <1 month. Anorectal manometry: absence of the rectoanal inhibitory reflex (RAIR) — useful as a screening tool but not diagnostic alone.
Surgical Treatment — Pull-Through Procedures
| Procedure | Technique | Key Features |
|---|---|---|
| Swenson | Full-thickness resection of aganglionic segment; colorectal/coloanal anastomosis | Original procedure; higher risk of pelvic nerve injury |
| Duhamel | Ganglionated bowel brought behind the aganglionic rectum; side-to-side anastomosis using a stapler | Preserves native rectum; risk of blind rectal pouch |
| Soave (endorectal) | Mucosectomy of the aganglionic rectum; ganglionated bowel pulled through the aganglionic muscular cuff | Most widely used; lower pelvic nerve injury risk; cuff abscess possible |
| Transanal (De la Torre-Mondragon) | Entirely transanal approach; submucosal dissection and pull-through | No abdominal incision; for short-segment disease; fastest recovery |
Most centers now perform a single-stage transanal endorectal pull-through (Soave variant) for short-segment disease in the neonatal period or early infancy, avoiding the need for a colostomy. Frozen section confirmation of ganglion cells at the proximal margin is mandatory.
11 Pyloric Stenosis
Hypertrophic pyloric stenosis (HPS) results from hypertrophy and hyperplasia of the pyloric muscle, causing gastric outlet obstruction. Incidence: 2-5 per 1,000 live births. M:F = 4-5:1. Typically presents at 3-6 weeks of age (rarely before 1 week or after 12 weeks). Risk factors: firstborn male, family history, erythromycin/azithromycin exposure in the first 2 weeks of life.
Clinical Presentation
Nonbilious, projectile vomiting immediately after feeds (the obstruction is proximal to the ampulla of Vater). The infant is hungry immediately after vomiting ("hungry vomiter"). Visible gastric peristaltic waves may be seen moving from left to right across the upper abdomen. An "olive" mass (the hypertrophied pylorus) is palpable in the right upper quadrant/epigastrium in 60-80% of experienced examiners — this finding alone is diagnostic.
Metabolic Derangement
Loss of gastric HCl causes chloride depletion and metabolic alkalosis. The kidneys initially compensate by excreting bicarbonate (alkaline urine). As volume depletion worsens, the kidneys prioritize sodium and water reabsorption — exchanging H+ and K+ for Na+ in the distal tubule, producing paradoxical aciduria (acidic urine despite systemic alkalosis) and worsening hypokalemia. This metabolic derangement must be corrected BEFORE surgery — the priority is resuscitation, not the OR.
Resuscitation protocol: NS bolus (20 mL/kg) for initial volume expansion. Then D5 0.45% NS + 20-40 mEq/L KCl at 1.5x maintenance rate. Check electrolytes every 4-6 hours. Goals before surgery: serum Cl >100 mEq/L, K >3.5 mEq/L, HCO3 <30 mEq/L, CO2 <30, urine output >1 mL/kg/hr. Typical resuscitation takes 12-48 hours. Do NOT add potassium until the infant has voided (confirm renal function).
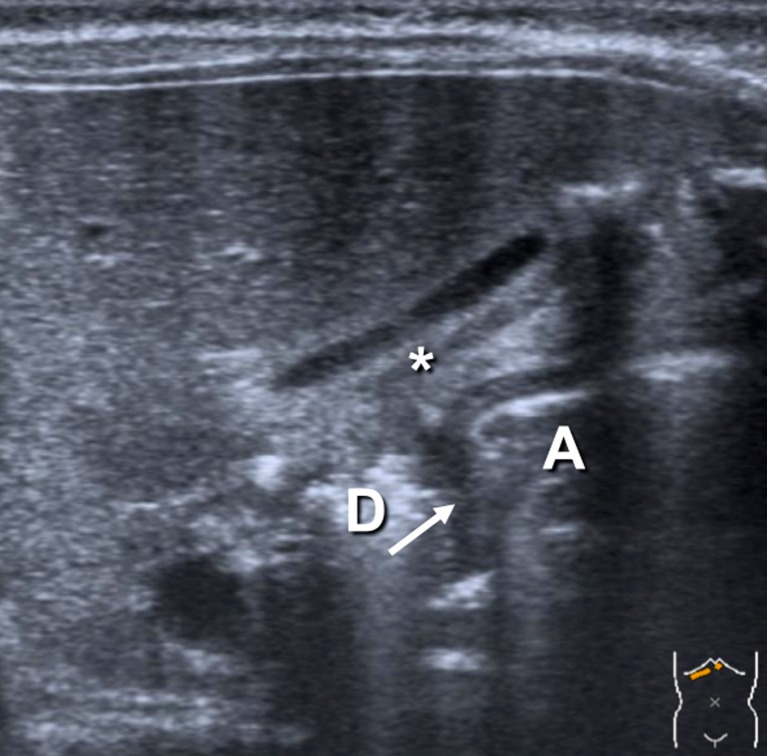
Diagnosis
Ultrasound is the gold standard. Diagnostic criteria: pyloric muscle thickness ≥3 mm (single wall), pyloric channel length ≥15 mm (the "3 and 15" rule, or alternatively "pi" mnemonic: pyloric index ≥14). Failure of relaxation and passage of fluid through the pylorus confirms the diagnosis.
Surgical Treatment — Ramstedt Pyloromyotomy
Described by Ramstedt in 1912. A longitudinal incision is made through the hypertrophied pyloric muscle (serosa and muscle only) without entering the mucosa. The incision extends from the pyloric vein of Mayo proximally to the gastric antrum. The muscle is spread bluntly until the mucosa bulges freely through the myotomy ("adequate split"). Completeness is confirmed by seeing the mucosa bulge independently on both sides of the myotomy and by passing a feeding catheter through the pylorus.
Approach: Traditionally through a right upper quadrant transverse incision (or periumbilical). Laparoscopic pyloromyotomy (typically using three 3 mm ports) is increasingly preferred — equivalent outcomes with improved cosmesis.
Complications: Incomplete myotomy (persistent vomiting — 1-2%; requires reoperation with completion myotomy at a different location on the pylorus). Duodenal or gastric mucosal perforation (1-4% — recognized intraoperatively by air leak test: inject air via NG tube while submerging the pylorus in saline; bubbles indicate a mucosal breach; repair with omental patch and interrupted sutures, then perform a new myotomy 90-180 degrees from the perforation site). Wound infection (1-5%, higher with open approach). Postoperative emesis (up to 70-80% in the first 24-48 hours) — this is normal and does not indicate incomplete myotomy; feeds can be advanced as tolerated. Most infants are discharged within 24 hours of surgery.
Postoperative Feeding Protocol
Ad libitum feeding (unrestricted feeds as tolerated by the infant) starting 4-6 hours postoperatively is the current standard — randomized trials show it is safe and shortens hospital stay compared to graduated feeding protocols. Parents should be counseled that some emesis is expected and does not indicate surgical failure. If vomiting persists beyond 5-7 days, obtain ultrasound to evaluate for incomplete myotomy.
12 Intussusception
Intussusception is the telescoping of a proximal segment of bowel (intussusceptum) into the distal segment (intussuscipiens). It is the most common cause of intestinal obstruction in children aged 6 months to 3 years (peak incidence 5-10 months). The most common type is ileocolic (terminal ileum telescopes into the cecum/ascending colon).
Etiology
In children <3 years: 90-95% are idiopathic — thought to be related to lymphoid hyperplasia (enlarged Peyer patches) serving as a lead point, often following a viral illness or rotavirus vaccination. In children >3 years: more likely to have a pathologic lead point — Meckel diverticulum, polyp, duplication cyst, lymphoma, Henoch-Schonlein purpura, or cystic fibrosis.
Clinical Presentation
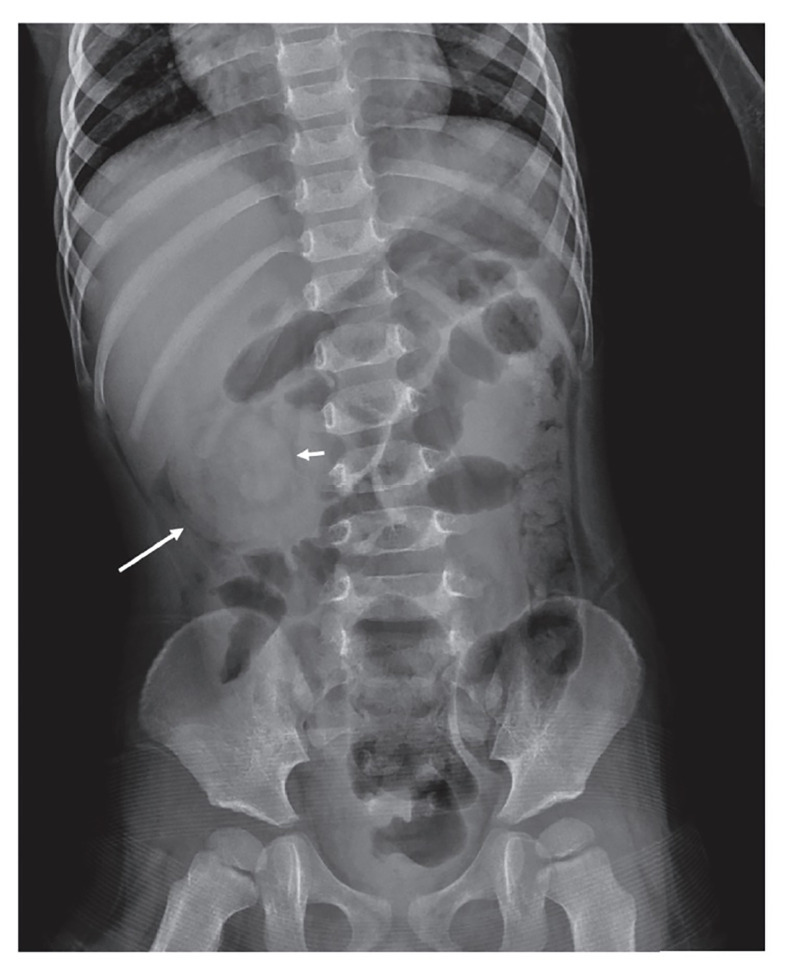
Classic triad (present in only ~20%): (1) episodic colicky abdominal pain (child draws knees up, inconsolable for 15-20 minutes, then appears well between episodes), (2) "currant jelly" stool (bloody mucus — a late and ominous sign indicating mucosal ischemia), (3) palpable sausage-shaped mass in the right upper quadrant or epigastrium. Lethargy may be the predominant presenting symptom and can mimic sepsis or neurologic disease.
Diagnosis
Ultrasound is the diagnostic study of choice: "target sign" or "doughnut sign" on transverse view (concentric rings of edematous bowel), "pseudokidney" sign on longitudinal view. Sensitivity and specificity both >98%. Abdominal X-ray may show a soft tissue mass, paucity of bowel gas in the right lower quadrant, or signs of obstruction — but is often normal early.
Nonoperative Reduction
Air enema reduction (pneumatic reduction) is the first-line treatment in most centers: air is insufflated per rectum under fluoroscopic guidance. Successful reduction is confirmed by free reflux of air into the terminal ileum. Success rate: 80-95%. Hydrostatic reduction (saline or contrast under ultrasound guidance) is an alternative. Maximum pressure: 120 mmHg for air enema. The "rule of 3s" is used by some: 3 attempts, 3 minutes each, with 3-minute intervals.
Contraindications to Nonoperative Reduction
Peritonitis, free air (perforation), hemodynamic instability, sepsis. Relative: prolonged symptoms (>48 hours), small bowel obstruction pattern on X-ray, age <3 months or >5 years (higher likelihood of lead point).
Surgical Management
Indications: failed enema reduction (after 3 attempts), perforation, peritonitis, recurrent intussusception (especially >3 episodes — search for lead point), or suspected lead point. Operative approach: manual reduction by gentle retrograde compression ("milking" the intussusceptum out — push, do not pull) through a right lower quadrant or periumbilical incision. If bowel is necrotic or a lead point is found, segmental resection with primary anastomosis. Appendectomy is routinely performed. Recurrence rate: 5-10% after nonoperative reduction, 1-3% after surgical reduction.
13 Inguinal Hernia, Umbilical Hernia & Meckel Diverticulum
Inguinal Hernia
Nearly all pediatric inguinal hernias are indirect — caused by a patent processus vaginalis (PPV). Incidence: 1-5% of term infants, 9-11% of premature infants. M:F = 6:1. Right-sided in 60% (later closure of the right processus vaginalis due to later testicular descent). Bilateral in 10-15%.
Incarceration risk: Highest in the first year of life — up to 30% in infants <6 months. An incarcerated hernia presents as a firm, tender, irreducible inguinal mass. Reduction is attempted with gentle sustained pressure (Taxis maneuver) after sedation and Trendelenburg positioning. If successful, repair is scheduled within 24-48 hours. If irreducible or signs of strangulation (erythema, edema, peritonitis), emergent exploration is required.
Surgical repair: Inguinal hernia repair is the most commonly performed operation in pediatric surgery. The standard approach is high ligation of the hernia sac at the internal ring — the processus vaginalis is identified, dissected from the cord structures, ligated at the internal ring, and excised. There is no need for a mesh repair or floor reconstruction (unlike adult hernias — the floor is normal in children). Laparoscopic repair is an alternative, with the advantage of allowing inspection of the contralateral side.
Umbilical Hernia
Extremely common: present in 10-30% of neonates, higher in African American infants (up to 75%) and premature infants. Most close spontaneously by age 4-5 years. Incarceration is exceedingly rare in children (unlike adults).
Indications for repair: (1) Persistent hernia after age 4-5 years, (2) defect >1.5-2 cm (unlikely to close spontaneously), (3) incarceration (rare), (4) symptoms. Repair: subumbilical incision, fascial closure (vest-over-pants or simple suture closure of the defect), preservation of the umbilical skin for cosmesis.
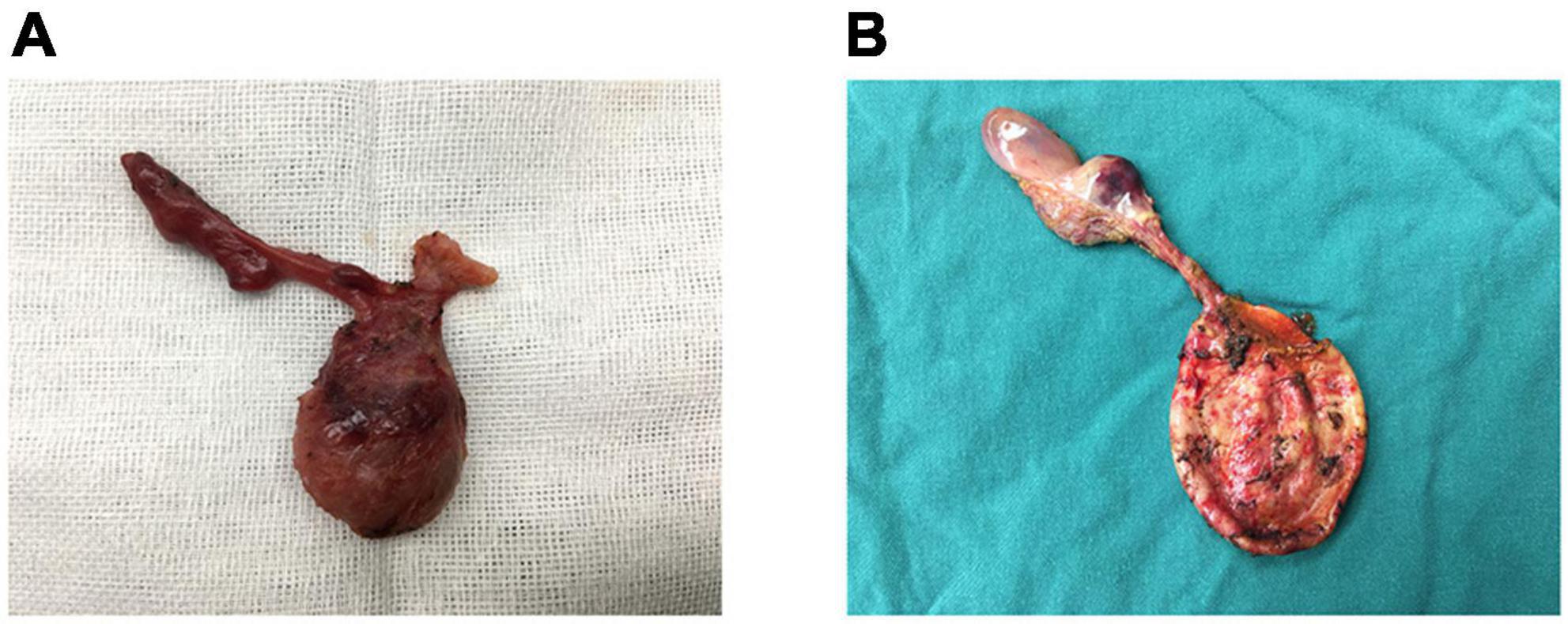
Meckel Diverticulum
The most common congenital anomaly of the GI tract. A true diverticulum (contains all bowel wall layers) representing a remnant of the omphalomesenteric (vitelline) duct.
2% of the population. 2 feet from the ileocecal valve. 2 inches long. 2 types of ectopic mucosa (gastric — most common, and pancreatic). 2 years of age (most common age at presentation). 2:1 male:female ratio.
Presentations: (1) Painless rectal bleeding (most common in children) — from ectopic gastric mucosa producing acid that ulcerates adjacent ileal mucosa. (2) Intestinal obstruction — from intussusception (Meckel as lead point), volvulus around a persistent vitelline band, or internal hernia. (3) Meckel diverticulitis — mimics appendicitis. (4) Perforation.
Diagnosis: Meckel scan (technetium-99m pertechnetate scintigraphy) — pertechnetate is taken up by ectopic gastric mucosa. Sensitivity 85-90% in children (lower in adults). Pretreatment with cimetidine (H2 blocker) or pentagastrin enhances uptake and sensitivity.
Treatment: Symptomatic Meckel — resection (diverticulectomy or segmental ileal resection with anastomosis if the base is wide or adjacent ileal ulceration is present). Incidental Meckel found at laparotomy — resection is controversial but generally recommended if the diverticulum has a narrow base, palpable heterotopic tissue, or the patient is young.
14 Necrotizing Enterocolitis
Necrotizing enterocolitis (NEC) is the most common surgical emergency in premature neonates. It primarily affects premature infants (90% of cases), with peak incidence at 30-32 weeks gestational age. The pathogenesis involves an immature intestinal barrier, bacterial translocation, and an exaggerated inflammatory response, often triggered by enteral feeding in the setting of prematurity. The terminal ileum and ascending colon are most commonly involved.
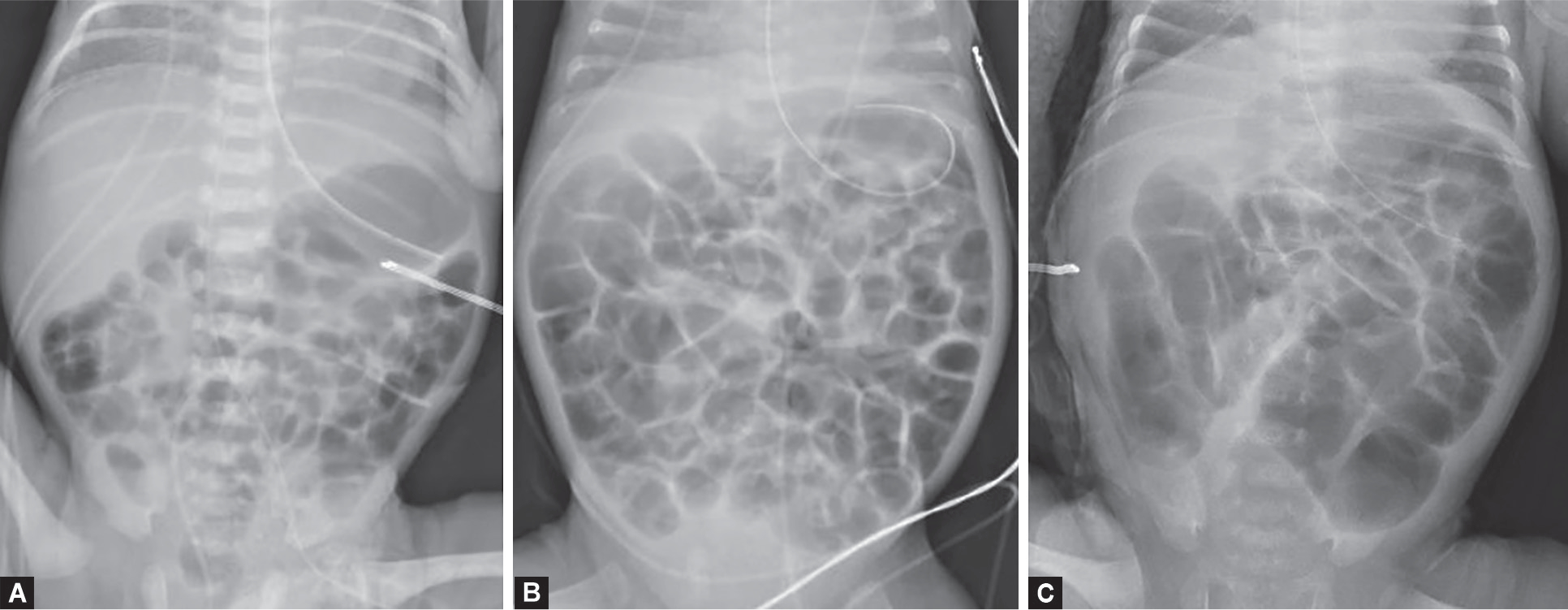
Modified Bell Staging
| Stage | Clinical | Radiographic | Management |
|---|---|---|---|
| IA — Suspected | Temperature instability, apnea, bradycardia, abdominal distension, gastric residuals, occult blood | Normal or mild ileus | NPO, antibiotics x 3 days, serial exams |
| IB — Suspected | Same as IA + gross bloody stool | Same as IA | Same as IA |
| IIA — Definite, mild | Same as I + absent bowel sounds, abdominal tenderness | Pneumatosis intestinalis (intramural gas — pathognomonic) | NPO, antibiotics x 7-14 days, serial exams q6-8h |
| IIB — Definite, moderate | Same as IIA + abdominal wall cellulitis, RLQ mass, metabolic acidosis, thrombocytopenia | Pneumatosis + portal venous gas, ascites | Same + consider surgical consultation |
| IIIA — Advanced, no perf | Worsening shock, DIC, severe metabolic acidosis, respiratory failure | Prominent ascites, dilated fixed loops | Surgery indicated: PPD or laparotomy |
| IIIB — Advanced, perforation | Same as IIIA + peritonitis, abdominal wall erythema | Pneumoperitoneum (free air) | Emergent surgery |
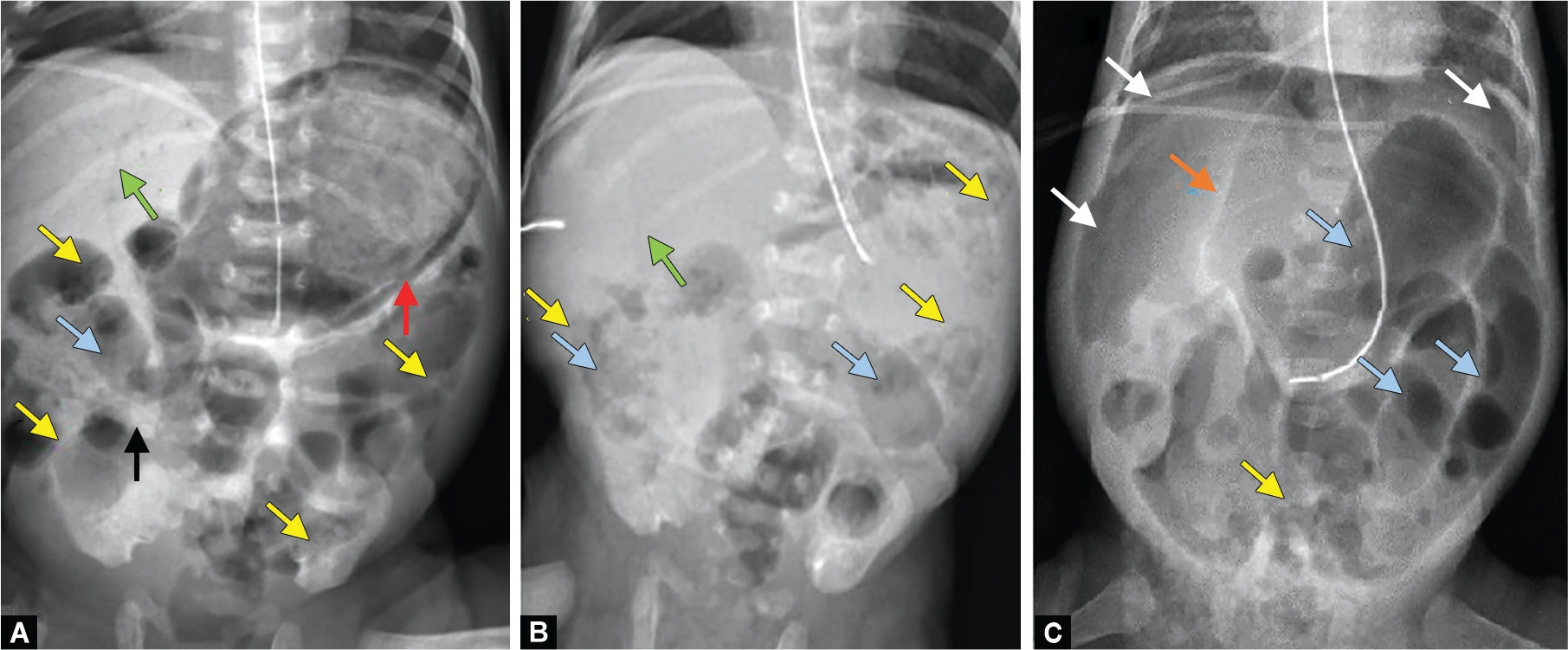
Indications for Surgery
Absolute: Pneumoperitoneum (free air — intestinal perforation). Relative: Clinical deterioration despite maximal medical therapy, portal venous gas, fixed dilated bowel loop on serial X-rays, abdominal wall erythema, palpable abdominal mass, worsening acidosis/thrombocytopenia, positive paracentesis (brown fluid, bacteria on Gram stain).
Surgical Options
Primary peritoneal drain (PPD): Bedside procedure for extremely low birth weight (<1000 g) or hemodynamically unstable neonates who may not survive transport to the OR. A Penrose drain is placed in the right lower quadrant under local anesthesia. May be definitive or a bridge to laparotomy. Laparotomy: Resection of necrotic bowel with stoma creation (enterostomy). Primary anastomosis is controversial in the acute setting. A "clip and drop back" technique may be used when viability is uncertain — clips mark margins, and a second look is performed in 48 hours.
15 Appendicitis in Children
Appendicitis is the most common surgical emergency in children overall (NEC is the most common in neonates). Peak incidence: 10-12 years of age. The diagnosis is more challenging in children than adults — perforation rates are higher at presentation (30-40% in children <5 years, up to 80-100% in children <3 years) due to atypical presentations and difficulty with history.
Pediatric Appendicitis Score (PAS — Samuel)
| Criterion | Score |
|---|---|
| Cough/hop/percussion tenderness | 2 |
| Anorexia | 1 |
| Fever ≥38°C | 1 |
| Nausea/vomiting | 1 |
| RLQ tenderness | 2 |
| Leukocytosis >10,000 | 1 |
| Neutrophilia (left shift, ANC >7,500) | 1 |
| Migration of pain to RLQ | 1 |
| Total: 10. Score ≤3 = low risk; 4-6 = equivocal (image); ≥7 = high risk (surgery) | |
Imaging
Ultrasound first — this is the recommended first-line imaging modality in children (avoiding radiation). Findings: noncompressible, blind-ending tubular structure >6 mm diameter, periappendiceal fluid, appendicolith. Sensitivity 85-95%, specificity 95-99% in experienced hands. If equivocal, follow with CT with IV contrast (no oral contrast needed) or MRI (increasing role, especially in adolescent females to exclude ovarian pathology). MRI has comparable accuracy to CT without radiation.
Management
Uncomplicated appendicitis: Laparoscopic appendectomy is the standard of care. Nonoperative management with antibiotics alone is an emerging approach (APPY trial, CODA trial) — success rates 65-75% at 1 year, with risk of recurrence. Operative management remains the primary recommendation.
Perforated appendicitis with abscess: Two approaches: (1) Interval appendectomy — initial IV antibiotics (piperacillin-tazobactam or ceftriaxone + metronidazole) +/- percutaneous CT-guided drainage of abscess >3 cm, followed by appendectomy 6-8 weeks later. Risk of recurrence without interval appendectomy is 15-20%. Some centers have moved away from routine interval appendectomy, reserving it for those with recurrent symptoms or concern for neoplasm (especially in older children/adolescents). (2) Early appendectomy — increasing evidence supports early laparoscopic surgery even with abscess, with equivalent outcomes, shorter total treatment time, and lower total cost, though with a higher complication rate at the index operation.
Antibiotic regimen: Uncomplicated appendicitis — preoperative dose only; no postoperative antibiotics needed (or 24 hours maximum). Perforated/complicated appendicitis — IV antibiotics for minimum 5 days, then transition to oral (amoxicillin-clavulanate) to complete 7-10 days total. Criteria for transition from IV to oral: afebrile for 24 hours, tolerating oral intake, WBC normalizing, no abdominal tenderness.
16 Biliary Atresia & Choledochal Cyst
Biliary Atresia
Progressive fibro-obliterative disease of the extrahepatic biliary tree, leading to cholestasis, biliary cirrhosis, and death if untreated. Incidence: 1 in 8,000-18,000 live births (higher in East Asia). It is the most common indication for liver transplantation in children.
Clinical presentation: Persistent conjugated (direct) hyperbilirubinemia beyond 2 weeks of age, acholic (pale/clay-colored) stools, dark urine, hepatomegaly. Any infant jaundiced beyond 2 weeks of age requires fractionation of bilirubin — an elevated direct bilirubin (>1 mg/dL or >20% of total) demands immediate workup.
Workup: Serum bilirubin fractionation, liver function tests (GGT is markedly elevated), abdominal ultrasound (absence of gallbladder or contracted/atretic gallbladder, "triangular cord" sign at the porta hepatis — echogenic triangular density >4 mm), hepatobiliary scintigraphy (HIDA scan — radiotracer is taken up by hepatocytes but not excreted into the intestine despite phenobarbital pretreatment), liver biopsy (bile duct proliferation, periportal fibrosis, bile plugs). Definitive diagnosis: intraoperative cholangiogram showing nonpatent extrahepatic biliary tree.
Kasai Portoenterostomy
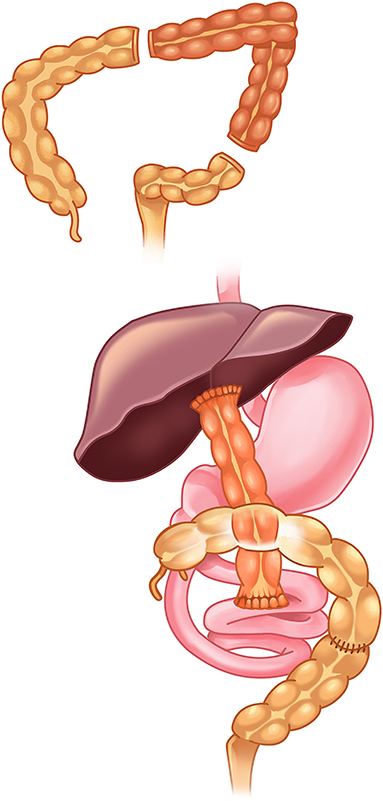
The Kasai procedure (hepatoportoenterostomy) is the initial surgical treatment. The fibrotic extrahepatic biliary remnant is excised at the porta hepatis (transecting at the level of the liver capsule to expose microscopic bile ductules), and a Roux-en-Y jejunal limb is anastomosed to the cut surface of the porta hepatis. The goal is to restore bile flow from the microscopic intrahepatic ductules through the surgically exposed portal plate into the Roux limb.
The Kasai procedure must be performed as early as possible — ideally before 60 days of age. Success rates (bile drainage achieved): >80% if performed before 30 days, ~50% at 60 days, <20% after 90 days. Even successful Kasai is not curative — progressive hepatic fibrosis continues, and up to 70-80% of patients will ultimately require liver transplantation. Post-Kasai cholangitis is common (40-60%) and must be treated aggressively with IV antibiotics.
Choledochal Cyst
Congenital cystic dilatation of the biliary tree. Incidence: 1 in 100,000-150,000 in Western countries (much higher in Japan: 1 in 1,000). F:M = 4:1.
Todani Classification
| Type | Description | Frequency |
|---|---|---|
| I | Fusiform dilatation of the common bile duct | 50-85% (most common) |
| II | True diverticulum of the CBD | 2-3% |
| III | Choledochocele (intraduodenal CBD dilatation) | 1-5% |
| IVa | Multiple intrahepatic and extrahepatic cysts | 15-35% |
| IVb | Multiple extrahepatic cysts only | Rare |
| V (Caroli disease) | Multiple intrahepatic cysts only | Rare |
Clinical triad (present in only 10-15%): RUQ pain, jaundice, palpable mass. More common presentations: intermittent jaundice, recurrent pancreatitis, cholangitis. Long-term cancer risk: choledochal cysts carry a significant risk of cholangiocarcinoma (up to 15-20% if untreated by adulthood). This risk mandates complete excision regardless of symptoms.
Treatment: Complete excision of the cyst with Roux-en-Y hepaticojejunostomy. The entire cyst wall must be removed to eliminate cancer risk. The gallbladder is removed (it is often connected to the cyst). Internal drainage procedures (cystoduodenostomy, cystoenterostomy) without excision are obsolete — they do not eliminate the malignant potential and have higher complication rates (cholangitis, stricture, cancer). Type III (choledochocele) may be managed with endoscopic sphincterotomy if small. Type IVa (intra- and extrahepatic): excision of extrahepatic component with hepaticojejunostomy; may require hepatectomy for severely affected hepatic segments. Type V (Caroli disease): partial hepatectomy if disease is limited to one lobe, liver transplantation if diffuse. Long-term surveillance with ultrasound and liver function tests is recommended even after complete excision.
17 Short Bowel Syndrome & Intestinal Duplications
Short Bowel Syndrome (SBS)
SBS results from massive intestinal resection or congenital short bowel, leaving insufficient absorptive surface to maintain nutrition via the enteral route alone. In neonates, the most common causes are NEC (massive bowel necrosis), midgut volvulus, and gastroschisis with extensive atresia. Defined as residual small bowel length <25% of expected for gestational age (approximately <75 cm in a term neonate).
Management — Intestinal Rehabilitation: The cornerstone is gradual advancement of enteral nutrition (trophic feeds, then slow increases) combined with TPN to meet caloric needs. An intestinal rehabilitation team (surgeon, gastroenterologist, dietitian, pharmacist) optimizes outcomes. Key medications: loperamide (reduces motility), cholestyramine (bile acid malabsorption), proton pump inhibitors (gastric hypersecretion), teduglutide (GLP-2 analogue — promotes intestinal adaptation by increasing villus height and crypt depth; FDA-approved for SBS).
Autologous Intestinal Reconstruction
| Procedure | Technique | Indication |
|---|---|---|
| Bianchi procedure (longitudinal intestinal lengthening and tailoring — LILT) | The dilated bowel is split longitudinally between the two leaves of the mesentery, creating two hemiloops, each with its own mesenteric blood supply, which are then anastomosed in series | Dilated bowel in SBS to double bowel length |
| STEP procedure (serial transverse enteroplasty) | Alternating stapler applications from opposite sides of the dilated bowel create a zigzag channel, lengthening the bowel without dividing the mesentery | Dilated bowel in SBS; technically simpler than Bianchi; can be repeated |
If intestinal rehabilitation and reconstruction fail, intestinal transplantation (isolated small bowel or multivisceral) is the definitive option. Indications: TPN failure (liver failure, loss of IV access sites, recurrent line sepsis), TPN-dependent with <10 cm of small bowel.
Intestinal Duplications
Spherical or tubular cystic structures that share a common muscular wall and blood supply with the adjacent bowel. They can occur anywhere along the GI tract (ileum is most common — 35%). Most present in the first 2 years of life with obstruction, bleeding (if ectopic gastric mucosa is present), or a palpable mass. Treatment: resection (often requiring resection of the adjacent bowel segment due to shared blood supply), or mucosal stripping for long tubular duplications where resection would sacrifice too much bowel.
Mesenteric & Omental Cysts
Rare cystic lesions within the mesentery or omentum, often lymphatic malformations (cystic lymphangioma). Present with abdominal distension, palpable mass, or acute abdomen (torsion, rupture, hemorrhage, intestinal obstruction from compression). Ultrasound/CT: well-defined cystic mass, often multiloculated, with septations and no solid components. Treatment: complete excision is the goal. If excision would require extensive bowel resection, marsupialization or partial excision is an alternative. Sclerotherapy (injection with OK-432 or doxycycline) may be used for large, unresectable lymphatic malformations. Recurrence rate is higher with incomplete excision.
Meconium Peritonitis
Sterile chemical peritonitis resulting from antenatal bowel perforation with meconium spillage. Causes: intestinal atresia, meconium ileus, volvulus, internal hernia. Prenatal: intra-abdominal calcifications on ultrasound (pathognomonic), ascites, pseudocyst formation. Postnatal: abdominal distension, bilious emesis, or may be asymptomatic if perforation has sealed. Abdominal X-ray: scattered calcifications throughout the abdomen. Treatment depends on the underlying cause: resection of the perforated segment, irrigation, primary anastomosis or stoma. Simple sealed perforations may not require surgery if the infant is otherwise well.
18 Congenital Lung Lesions
Congenital Pulmonary Airway Malformation (CPAM/CCAM)
Hamartomatous lesion of the lung with cystic and/or solid components, resulting from abnormal branching morphogenesis. Most diagnosed prenatally on ultrasound. The Stocker classification defines subtypes:
| Type | Features | Frequency |
|---|---|---|
| Type 0 | Acinar dysplasia (incompatible with life) | 1-3% |
| Type 1 | Large cysts (2-10 cm); most common; excellent prognosis; risk of bronchoalveolar carcinoma | 60-70% |
| Type 2 | Small cysts (0.5-2 cm); associated with other anomalies (renal, cardiac); intermediate prognosis | 15-20% |
| Type 3 | Solid/microcystic (adenomatoid); large lesions causing mediastinal shift; associated with hydrops | 5-10% |
| Type 4 | Large thin-walled peripheral cysts; risk of pleuropulmonary blastoma | Rare |
Management: Asymptomatic prenatally detected lesions: serial imaging. If hydrops develops in utero, fetal intervention may be required (thoracoamniotic shunt for large cysts, EXIT procedure, or fetal resection for solid lesions). Postnatally: elective lobectomy at 3-6 months of age is recommended even for asymptomatic lesions due to risk of recurrent infection and malignant transformation (Type 1 — bronchoalveolar carcinoma; Type 4 — pleuropulmonary blastoma). Surgical approach: thoracoscopic or open lobectomy.
Bronchopulmonary Sequestration (BPS)
Mass of nonfunctioning lung tissue that receives arterial blood supply from the systemic circulation (typically an aberrant artery from the aorta) rather than the pulmonary artery. No connection with the normal tracheobronchial tree.
| Feature | Intralobar (75%) | Extralobar (25%) |
|---|---|---|
| Pleura | Shares pleura with normal lung | Has own pleural envelope |
| Venous drainage | Pulmonary veins (L-to-L shunt) | Systemic veins (IVC, azygos, hemiazygos) |
| Location | Left lower lobe (60%) | Left lower lobe (90%), can be subdiaphragmatic |
| Presentation | Recurrent pneumonia | Often incidental, respiratory distress in neonates |
| Associated anomalies | Rare | Common (CDH, cardiac anomalies) |
| Treatment | Lobectomy | Excision (does not require lobectomy) |
Congenital Lobar Emphysema (CLE)
Progressive hyperinflation of one or more lobes due to a ball-valve mechanism (cartilage deficiency, bronchial obstruction, extrinsic compression). Most commonly affects the left upper lobe (43%), followed by the right middle lobe (32%) and right upper lobe (20%). Presentation: progressive respiratory distress in the first weeks to months of life. CXR: hyperlucent, hyperexpanded lobe with compression of adjacent lung and mediastinal shift. Treatment: lobectomy for symptomatic patients. Asymptomatic patients may be observed as some cases resolve spontaneously.
Foreign Body Aspiration
Peak incidence 1-3 years. Most common foreign body: peanuts and other food items. Right mainstem bronchus is more commonly affected (wider, more vertical angle). Presentation: acute choking episode, followed by cough, wheezing, unilateral decreased breath sounds. Three phases: (1) initial choking/gagging, (2) asymptomatic interval (hours to weeks — may lead to delayed diagnosis), (3) complications (pneumonia, atelectasis, abscess). CXR may show hyperinflation of the affected side (ball-valve air trapping — more apparent on expiratory film or bilateral decubitus views) or atelectasis. Radiopaque objects are visible; organic matter (peanuts) is not.
Treatment: Rigid bronchoscopy under general anesthesia with spontaneous ventilation — the gold standard for diagnosis and extraction. Optical forceps, balloon catheters, and basket retrievers are used depending on the object. Flexible bronchoscopy is used for diagnosis in some centers but rigid bronchoscopy provides better airway control and instrument access. Post-extraction: observe for laryngeal edema, repeat CXR, short course of dexamethasone if significant airway edema.
19 Chest Wall Deformities & Mediastinal Masses
Pectus Excavatum
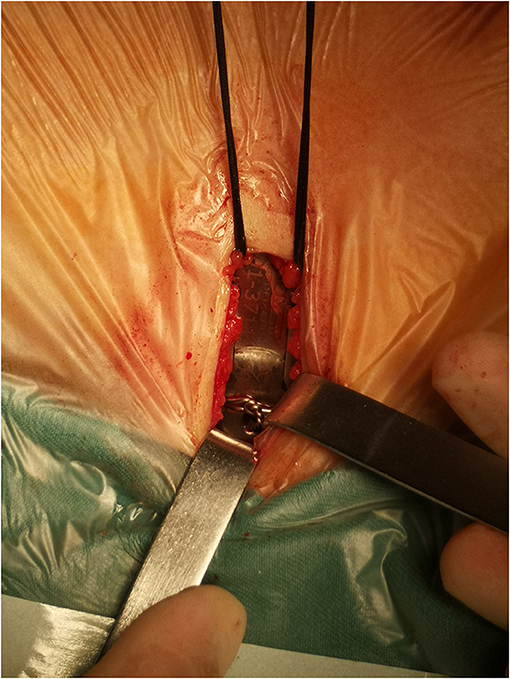
Depression of the sternum ("funnel chest") — the most common chest wall deformity in children (1 in 300-400). M:F = 3:1. Often worsens during pubertal growth spurt. Severity assessed by the Haller index (CT measurement: transverse diameter / AP diameter at the deepest point of depression; normal ~2.5; ≥3.25 = severe, surgical indication).
Nuss procedure (minimally invasive repair): A curved steel bar is inserted through bilateral thoracic incisions, passed behind the sternum under thoracoscopic guidance, and flipped to push the sternum anteriorly. The bar remains in place for 2-3 years, then is removed. Complications: bar displacement, pneumothorax, pericardial injury.
Ravitch procedure (open repair): Subperichondrial resection of the deformed costal cartilages with sternal osteotomy and fixation. More invasive but provides direct correction. Generally reserved for recurrent or complex deformities.
Pectus Carinatum
Protrusion of the sternum ("pigeon chest"). Less common than excavatum (ratio 1:5). Two subtypes: chondrogladiolar (most common — protrusion of the body of the sternum) and chondromanubrial (upper sternum — "Pouter pigeon," more difficult to treat). Primarily a cosmetic concern but may cause chest pain with exertion and exercise intolerance. First-line treatment: external bracing (dynamic compression brace worn 14-23 hours/day for 6-18 months — effective in 70-80% of compliant patients; most effective when started before skeletal maturity). Surgery (modified Ravitch with subperichondrial cartilage resection and sternal osteotomy) reserved for bracing failure, rigid deformities in older patients, or severe asymmetric variants.
Mediastinal Masses in Children
| Compartment | Common Tumors | Key Features |
|---|---|---|
| Anterior | Lymphoma (Hodgkin and non-Hodgkin), teratoma/germ cell tumor, thymic lesions | Risk of airway compression — assess before sedation/anesthesia |
| Middle | Lymphoma, bronchogenic cyst, granuloma | Bronchial/vascular compression possible |
| Posterior | Neuroblastoma, ganglioneuroma, neurofibroma | Neural origin; may extend into spinal canal (dumbbell tumors) |
An anterior mediastinal mass in a child can cause life-threatening airway compression, especially under general anesthesia (loss of muscle tone allows the mass to further compress the trachea). Before ANY sedation or general anesthesia, obtain a CT to assess tracheal compression. If >50% tracheal narrowing, avoid general anesthesia if possible — perform biopsy under local anesthesia (lymph node biopsy, bone marrow, pleural fluid). Consider empiric steroids for lymphoma if tissue diagnosis cannot be safely obtained.
20 Neuroblastoma
Neuroblastoma is the most common extracranial solid tumor in children, arising from neural crest cells. Median age at diagnosis: 17 months. Most common locations: adrenal gland (40%), paraspinal sympathetic chain (25%), posterior mediastinum (15%). Can occur anywhere along the sympathetic chain.
Clinical Presentation
Presentation varies by location. Abdominal (most common): firm, nodular mass crossing midline — unlike Wilms tumor which rarely crosses midline. Periorbital ecchymosis ("raccoon eyes") — from orbital metastases. Opsoclonus-myoclonus syndrome ("dancing eyes, dancing feet") — paraneoplastic chaotic eye movements with myoclonus; paradoxically associated with favorable tumor biology but neurologic sequelae persist. Horner syndrome (ptosis, miosis, anhidrosis) — cervical/thoracic tumors on sympathetic chain. Watery diarrhea (VIP secretion). Hypertension/tachycardia (catecholamine secretion). "Blueberry muffin baby" — subcutaneous metastatic nodules in neonates (Stage MS). Spinal cord compression from dumbbell tumors extending through neural foramina.
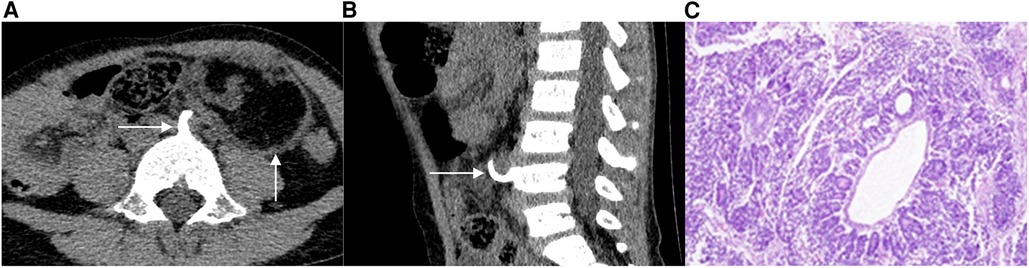
Tumor markers and imaging: Urine VMA and HVA elevated in >90% (spot urine, creatinine-normalized). Serum LDH, ferritin, and NSE (neuron-specific enolase) are prognostic. MIBG scan (meta-iodobenzylguanidine): highly specific for neuroblastoma, taken up by catecholamine-producing cells — used for staging and surveillance. CT/MRI for primary tumor assessment. Bone marrow biopsy from bilateral iliac crests (for staging — >10% marrow involvement upgrades to Stage M).
International Neuroblastoma Risk Group Staging System (INRGSS)
| Stage | Description |
|---|---|
| L1 | Localized, no image-defined risk factors (IDRFs) |
| L2 | Localized with one or more IDRFs (encasing vessels, infiltrating adjacent organs) |
| M | Distant metastatic disease |
| MS | Metastatic disease in children <18 months with metastases limited to skin, liver, and/or bone marrow (<10% involvement) — formerly Stage 4S; excellent prognosis; may spontaneously regress |
Risk Stratification
Based on age, stage, histology (Shimada classification), MYCN amplification (the single most important adverse prognostic factor), ploidy, and segmental chromosomal alterations. Low risk: observation or surgery alone (survival >95%). Intermediate risk: surgery + moderate chemotherapy (survival ~90%). High risk: intensive multimodal therapy — induction chemotherapy, surgery, high-dose chemotherapy with autologous stem cell rescue, radiation, anti-GD2 immunotherapy (dinutuximab), isotretinoin (13-cis-retinoic acid for differentiation therapy). Survival ~50%.
21 Wilms Tumor (Nephroblastoma)
Wilms tumor is the most common renal malignancy in children. Peak incidence: 3-4 years. Incidence: 1 in 10,000 children. Bilateral in 5-7%. Associated with syndromes: WAGR (Wilms, Aniridia, GU anomalies, intellectual disability — 11p13 deletion including WT1), Beckwith-Wiedemann (hemihypertrophy, macroglossia — 11p15.5), Denys-Drash (nephropathy, male pseudohermaphroditism — WT1 mutation).
Clinical Presentation
Asymptomatic abdominal mass discovered by parent or during routine examination — the classic presentation. The mass is usually smooth, firm, and does not cross the midline (unlike neuroblastoma). Other features: hematuria (25%), hypertension (25% — renin secretion), fever, abdominal pain. Do not aggressively palpate a suspected Wilms tumor — risk of tumor rupture and upstaging.
COG (Children's Oncology Group) Staging
| Stage | Description |
|---|---|
| I | Tumor limited to the kidney, completely excised; renal capsule intact |
| II | Tumor extends beyond kidney but completely excised (renal vein, perirenal tissue) |
| III | Residual tumor confined to abdomen (positive margins, lymph nodes, peritoneal spillage, biopsy before resection) |
| IV | Hematogenous metastases (lung, liver, bone, brain) |
| V | Bilateral disease at diagnosis |
Treatment
North American approach (COG/NWTS): Upfront radical nephrectomy through a generous transverse or chevron transabdominal incision. Key steps: early ligation of the renal vein and artery (before tumor manipulation to prevent hematogenous spread), wide Gerota fascia excision, lymph node sampling from the renal hilum and para-aortic/paracaval region, and inspection of the contralateral kidney. Favorable histology (90% of cases): vincristine + dactinomycin (Stages I-II), with doxorubicin added for Stage III-IV. Anaplastic histology (unfavorable — 10%): more aggressive regimens including cyclophosphamide, carboplatin, etoposide.
European approach (SIOP): Preoperative chemotherapy (to reduce tumor size and risk of intraoperative rupture) followed by surgery. Both approaches yield comparable survival (~90% overall for favorable histology).
Bilateral Wilms (Stage V): Preoperative chemotherapy to shrink tumors (6-12 weeks), then bilateral nephron-sparing surgery (partial nephrectomy of both kidneys) to preserve maximal renal parenchyma. Imaging is repeated after chemotherapy to assess response and plan surgery. Complete nephrectomy of one side with partial of the other is an alternative if nephron-sparing is not possible bilaterally. The goal is to avoid bilateral nephrectomy and dialysis/transplantation whenever possible.
Prognosis: Overall survival for favorable histology Wilms tumor: Stage I/II >95%, Stage III ~90%, Stage IV ~85%. Anaplastic (unfavorable) histology has significantly worse outcomes: diffuse anaplasia ~55% survival. Late effects of treatment include cardiotoxicity (doxorubicin), renal insufficiency (especially bilateral disease), second malignancies, and musculoskeletal effects from radiation.
22 Hepatoblastoma, Rhabdomyosarcoma & Sacrococcygeal Teratoma
Hepatoblastoma
Most common primary liver malignancy in children. Peak incidence: <3 years. Associated with Beckwith-Wiedemann syndrome, familial adenomatous polyposis (FAP), and extreme prematurity. Tumor marker: alpha-fetoprotein (AFP) — markedly elevated in >90%; used for diagnosis and monitoring treatment response.
PRETEXT Staging (PRE-Treatment EXTent of disease)
Based on preoperative imaging, dividing the liver into 4 sections (left lateral, left medial, right anterior, right posterior):
| PRETEXT | Description |
|---|---|
| I | Tumor involves 1 section; 3 contiguous sections are tumor-free |
| II | Tumor involves 1-2 sections; 2 contiguous sections are tumor-free |
| III | Tumor involves 2-3 sections; only 1 section or no contiguous sections tumor-free |
| IV | Tumor involves all 4 sections |
Treatment: Neoadjuvant chemotherapy (cisplatin + doxorubicin — most common regimen) to downstage the tumor, followed by surgical resection. Complete resection is critical for cure. If the tumor is unresectable after chemotherapy, liver transplantation is indicated (5-year survival 70-80% post-transplant for hepatoblastoma). AFP should normalize after successful treatment — persistent elevation suggests residual disease.
Rhabdomyosarcoma (RMS)
Most common soft tissue sarcoma in children. Arises from primitive mesenchymal cells (skeletal muscle lineage). Two main histologic subtypes: embryonal (most common, including botryoid variant — favorable prognosis) and alveolar (associated with PAX3-FOXO1 or PAX7-FOXO1 translocations — unfavorable prognosis). Common sites: head/neck (40%), genitourinary (25%), extremities (20%).
Treatment: Multimodal — incisional biopsy for diagnosis and staging (imaging with CT/MRI, bone marrow biopsy, bone scan, PET/CT). Chemotherapy is the backbone of treatment (vincristine, actinomycin D, cyclophosphamide — VAC regimen; irinotecan added for intermediate/high risk). Complete surgical resection is performed when feasible without functional mutilation — re-excision of positive margins is recommended. For orbital, parameningeal, and bladder/prostate RMS, chemotherapy + radiation is preferred over radical surgery to preserve function. Radiation therapy (41-50 Gy) for microscopic or gross residual disease. Clinical group staging (IRS): Group I = complete resection; Group II = microscopic residual; Group III = gross residual or biopsy only; Group IV = metastatic. Prognosis: embryonal favorable (85-90% survival); alveolar and metastatic disease much worse (25-30% survival).
Sacrococcygeal Teratoma (SCT)
Most common tumor in neonates. F:M = 4:1. Arises from totipotent cells at the coccyx (Hensen node). Contains tissue from all three germ layers.

Altman Classification
| Type | Description | Frequency | Malignancy Risk |
|---|---|---|---|
| I | Predominantly external, minimal presacral component | 47% | Low (8%) |
| II | External with significant intrapelvic component | 34% | Moderate (21%) |
| III | External with predominant pelvic/abdominal component | 9% | High (34%) |
| IV | Entirely presacral, no external component | 10% | Highest (38%) |
Treatment: Early resection including the coccyx (must remove the coccyx to prevent recurrence — failure to excise the coccyx is the most common cause of recurrence). Surgical approach: Types I-II — sacral/perineal approach; Types III-IV — combined abdominal and sacral approach. For neonates (predominantly mature teratoma): surgery alone is usually curative. For malignant SCT (yolk sac tumor component — immature elements or endodermal sinus tumor): neoadjuvant chemotherapy (cisplatin, etoposide, bleomycin — PEB regimen) followed by resection. AFP monitoring: elevated AFP is normal in neonates (physiologic — can be >100,000 ng/mL at birth) — must follow the age-appropriate decline curve (normalizes by 8-9 months of age); failure to decline or subsequent rise indicates malignancy or recurrence. Long-term follow-up: serial AFP levels and rectal examination every 3 months for 3 years, as recurrence may be presacral and clinically silent.
23 Ovarian Tumors in Children
Ovarian masses in children differ from adults — the majority are benign. The most common ovarian tumor in children is mature cystic teratoma (dermoid cyst), followed by functional/physiologic cysts.
Classification
| Category | Types | Key Features |
|---|---|---|
| Germ cell tumors (60-70%) | Mature teratoma, immature teratoma, yolk sac tumor (endodermal sinus tumor), dysgerminoma, embryonal carcinoma, choriocarcinoma | AFP elevated in yolk sac tumor; beta-hCG in choriocarcinoma; LDH in dysgerminoma |
| Epithelial tumors (10-15%) | Cystadenoma, cystadenocarcinoma | More common in adolescents; behave similarly to adult epithelial tumors |
| Sex cord-stromal (5-8%) | Granulosa cell tumor, Sertoli-Leydig cell tumor | May produce hormones (estrogen → precocious puberty; androgens → virilization) |
Surgical principles: Ovarian-sparing surgery whenever possible in children and adolescents to preserve fertility. Cystectomy (removal of the cyst with preservation of normal ovarian tissue) is appropriate for benign-appearing cysts (simple, thin-walled, no solid components, normal tumor markers). Use an endoscopic bag to avoid spillage. If malignancy is suspected (solid components, elevated tumor markers, peritoneal implants, Doppler flow within solid components), unilateral salpingo-oophorectomy with full surgical staging is performed: peritoneal washings, omental biopsy, peritoneal biopsies, ipsilateral pelvic and para-aortic lymph node sampling. Avoid rupture of the cyst during removal — spillage of a malignant cyst upstages the tumor. The contralateral ovary is inspected but not biopsied if grossly normal (risk of adhesions and compromising fertility).
Immature teratoma: Graded 0-3 based on amount of immature (neural) tissue. Grade 1: surgery alone is curative. Grade 2-3 or recurrent: chemotherapy (PEB — cisplatin, etoposide, bleomycin). Even malignant germ cell tumors have excellent prognosis in children with chemotherapy: >90% survival for early-stage disease.
24 Undescended Testis & Testicular Torsion
Undescended Testis (Cryptorchidism)
The most common genitourinary anomaly in boys. Incidence: 3% of term males at birth (30% in premature males), decreasing to 1% by 6 months (spontaneous descent). Bilateral in 10-20%. Right side more common. A truly undescended testis is one that cannot be manually brought into the scrotum and remain there.
Classification: Palpable (80%) — inguinal canal, prescrotal, or ectopic (superficial inguinal pouch, femoral, perineal, contralateral). Nonpalpable (20%) — intra-abdominal, absent (vanishing testis from prenatal torsion), or atrophic.
Risks of undescended testis: Infertility (even unilateral — impaired spermatogenesis from elevated temperature), malignancy (4-10x increased risk of testicular cancer — seminoma most common; risk persists even after orchiopexy, though early surgery may reduce risk), testicular torsion, inguinal hernia (90% have a patent processus vaginalis), psychological impact.
Timing of orchiopexy: 6-12 months of age (AUA/AAP guidelines). Early surgery optimizes fertility potential and may reduce malignancy risk. If spontaneous descent has not occurred by 6 months, it will not occur.
Surgical approach: Palpable testis: inguinal orchiopexy (inguinal incision, mobilize the spermatic cord, divide the cremasteric fibers and processus vaginalis, create a subdartos pouch in the scrotum, fix the testis). Nonpalpable testis: diagnostic laparoscopy first — if viable intra-abdominal testis is found, Fowler-Stephens orchiopexy (two-stage): Stage 1 — ligate the spermatic (testicular) vessels laparoscopically, allowing collateral blood supply from the vas deferens (deferential artery) and cremasteric artery to develop over 6 months; Stage 2 — bring the testis into the scrotum on the collateral blood supply. If a blind-ending vas and vessels are found, the testis is absent (vanishing testis), and no further surgery is needed.
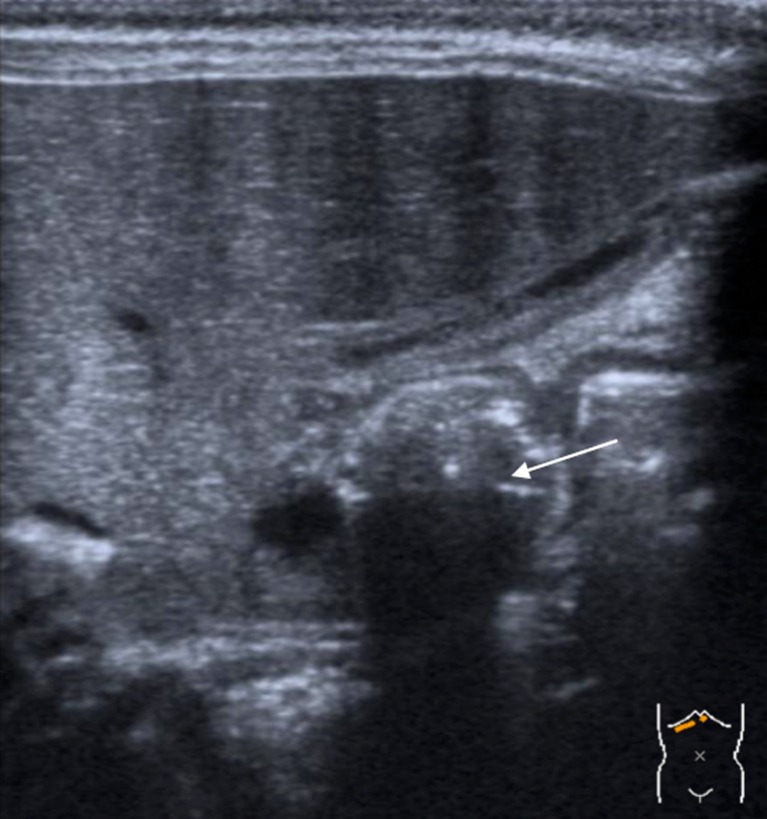
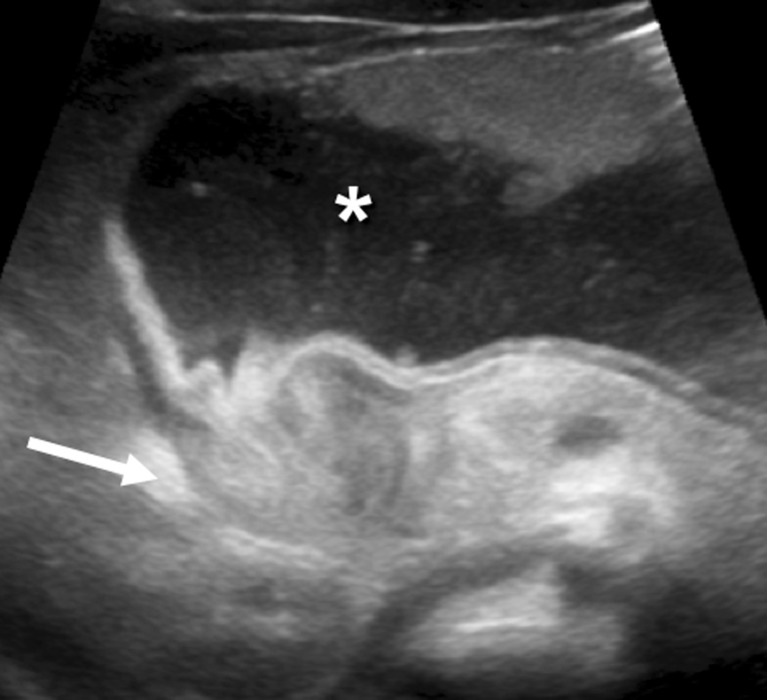
Testicular Torsion
Testicular torsion is a time-sensitive surgical emergency. The spermatic cord twists, occluding venous outflow and then arterial inflow. Testicular salvage rates: >90% if detorsion within 6 hours, ~50% at 12 hours, <10% after 24 hours. Do not delay surgery for imaging if the clinical picture is consistent.
Bimodal age distribution: Neonatal (extravaginal — the entire tunica vaginalis twists on the spermatic cord; often presents as a painless, hard, discolored scrotal mass at birth; salvage is rare) and adolescent (intravaginal — the testis twists within the tunica vaginalis; peak 12-18 years).
Bell clapper deformity: The tunica vaginalis inserts high on the spermatic cord, leaving the testis free to rotate within ("like a clapper in a bell"). Present bilaterally — hence the need for bilateral orchiopexy.
Clinical: Sudden-onset severe scrotal pain, nausea/vomiting, high-riding testis with transverse lie, absent cremasteric reflex (most sensitive clinical finding — absent in >95% of torsion). Ultrasound with Doppler: absent or decreased blood flow to the affected testis — but if clinical suspicion is high, go directly to surgery.
Surgery: Scrotal or inguinal incision, detorse the testis, wrap in warm saline gauze for 15-20 minutes, assess viability (color return, Doppler flow, bleeding from cut edge of tunica albuginea). If viable: bilateral orchiopexy (fix both testes to prevent recurrence — the bell clapper is bilateral). Three-point fixation with nonabsorbable sutures through the tunica albuginea to the dartos fascia. If nonviable: orchiectomy + contralateral orchiopexy.
25 Hydrocele, Hypospadias & Vesicoureteral Reflux
Hydrocele
Fluid collection within the tunica vaginalis around the testis. Two types:
| Type | Mechanism | Transillumination | Management |
|---|---|---|---|
| Communicating | Patent processus vaginalis allowing peritoneal fluid to flow in and out; fluctuates in size (larger when upright) | Positive | Surgical repair (inguinal approach, ligate PPV) — same as hernia repair; timing: after age 12-18 months |
| Non-communicating | Processus vaginalis is closed; residual fluid is trapped; fixed size | Positive | Observation — most resolve by 12-18 months as fluid resorbs; surgery if persistent >18 months or enlarging |
Hypospadias
Urethral meatus opens on the ventral (underside) surface of the penis. Incidence: 1 in 200-300 males. Classified by meatal position: distal/anterior (glanular, coronal, subcoronal — 70%), middle (midshaft — 15%), proximal/posterior (penoscrotal, scrotal, perineal — 15%). Associated findings: ventral curvature (chordee) and hooded foreskin (dorsal preputial hood). Do not circumcise — the foreskin is used for surgical reconstruction.
Surgical repair: Typically performed at 6-18 months of age. The TIP (tubularized incised plate) or Snodgrass procedure is the most commonly used technique for distal and midshaft hypospadias: the urethral plate is incised in the midline (relaxing incision), then tubularized over a stent, creating a neourethra. A dartos flap (from the foreskin) is used as a waterproofing layer over the repair. For proximal hypospadias with severe chordee, staged repairs (Bracka — first stage: chordee correction with inner preputial or buccal mucosal graft placement; second stage: tubularization) may be needed.
Complications: Urethrocutaneous fistula (most common — 5-15%), meatal stenosis, urethral stricture, glans dehiscence, diverticulum.
Vesicoureteral Reflux (VUR)
Retrograde flow of urine from the bladder into the ureter/kidney. The most common urologic condition in children. Incidence: 1% of children, 30-40% of children presenting with UTI. F:M = 5:1 in older children (M:F = 2:1 in neonates detected prenatally). Caused by a short intramural tunnel of the ureter through the bladder wall (normally the tunnel length-to-diameter ratio is 5:1; in VUR this ratio is reduced).
International Grading System
| Grade | Description |
|---|---|
| I | Reflux into ureter only (no dilatation) |
| II | Reflux into ureter, pelvis, calyces (no dilatation) |
| III | Mild ureteral and pelvic dilatation; mild blunting of fornices |
| IV | Moderate ureteral dilatation and tortuosity; moderate pelvic dilatation; blunting of fornices |
| V | Severe dilatation and tortuosity; loss of papillary impressions; intraparenchymal reflux |
Management: Grades I-III: conservative — antibiotic prophylaxis (TMP-SMX or nitrofurantoin), serial VCUG/ultrasound monitoring. Spontaneous resolution rate: 80% for Grades I-II, 50% for Grade III. Grades IV-V or breakthrough UTIs: surgical intervention. STING/Deflux procedure (subureteral injection): endoscopic injection of dextranomer/hyaluronic acid copolymer (Deflux) beneath the ureteral orifice to create a valve mechanism — success rate ~80% for low-grade VUR. Ureteral reimplantation (Cohen cross-trigonal or Politano-Leadbetter): open surgical creation of a longer submucosal tunnel — success rate >95%.
26 Posterior Urethral Valves & Ovarian Torsion
Posterior Urethral Valves (PUV)
The most common cause of congenital lower urinary tract obstruction in males. Incidence: 1 in 5,000-8,000 male births. Obstructing mucosal folds in the posterior urethra (between the verumontanum and the external sphincter) cause bladder outlet obstruction, leading to bladder hypertrophy, hydroureteronephrosis, and varying degrees of renal dysplasia.
Presentation: Prenatal: bilateral hydroureteronephrosis, dilated thick-walled bladder ("keyhole sign" on ultrasound), oligohydramnios (severe obstruction leads to decreased urine output → reduced amniotic fluid → pulmonary hypoplasia). Postnatal: weak urinary stream, palpable distended bladder, UTI, failure to thrive, uremia. Diagnosed by voiding cystourethrogram (VCUG) — dilated posterior urethra with abrupt caliber change at the level of the valves.
Treatment: Initial: bladder drainage with a small (5-8 Fr) feeding tube placed per urethra — do NOT use a Foley catheter (balloon can obstruct the bladder neck or ureteral orifices). If urethral catheter cannot be passed, suprapubic cystostomy. Correct electrolyte abnormalities (post-obstructive diuresis is common — replace fluids mL-for-mL with 0.45% NS + K+ as needed). Definitive: primary valve ablation — endoscopic incision/fulguration of the valves at the 5, 7, and 12 o'clock positions using a cold knife, Bugbee electrode, or holmium laser (avoid electrocautery near the external sphincter to preserve continence). In very small neonates (<2 kg or urethra too small for a cystoscope), a cutaneous vesicostomy (Blocksom technique) is performed — the dome of the bladder is brought to the skin between the umbilicus and pubic symphysis. Valve ablation is performed when the child grows to an appropriate size (typically 6-12 months). High diversion (ureterostomy or pyelostomy) is reserved for severe upper tract damage or persistent infection despite vesicostomy.
Long-term outcomes: Despite early treatment, up to 30-40% develop end-stage renal disease by age 10-20 years (renal dysplasia from in-utero damage is irreversible). "Valve bladder syndrome" — the bladder may remain poorly compliant and small despite valve ablation, requiring anticholinergics (oxybutynin), clean intermittent catheterization (CIC), and in some cases bladder augmentation (enterocystoplasty). Lifelong nephrology and urology follow-up is essential.
Ovarian Torsion
Ovarian torsion can occur at any age, including neonates. The ovary (often with an associated cyst or mass) twists on its vascular pedicle, causing venous congestion and eventual arterial compromise. Most common in prepubertal and adolescent girls. Presentation: sudden onset of severe lower abdominal/pelvic pain, often with nausea and vomiting. Ultrasound with Doppler: enlarged ovary with decreased or absent blood flow (though Doppler may be normal in early torsion — a normal Doppler does not exclude torsion).
Treatment: Emergent surgical exploration (laparoscopic preferred). Ovarian-sparing (detorsion) is the standard approach in children and adolescents, even if the ovary appears dusky or black — the ovary has remarkable recovery potential. Allow 10-15 minutes of warm perfusion after detorsion to assess reperfusion. Oophoropexy (fixing the ovary to the pelvic sidewall or utero-ovarian ligament to prevent recurrence) is controversial but often performed. Oophorectomy is reserved for clearly necrotic or malignant-appearing ovaries. Cystectomy of the underlying cyst is performed to remove the lead point if feasible.
Neonatal ovarian torsion: A special entity — often presents as an abdominal or pelvic cystic mass on prenatal ultrasound that becomes complex/heterogeneous (indicating torsion and hemorrhage). Management is controversial: some advocate early surgery (laparoscopic detorsion/cystectomy) to salvage the ovary; others accept observation with oophorectomy only if symptomatic, as most neonatal ovarian cysts are follicular and resolve spontaneously. Aspiration of simple cysts >4 cm may prevent torsion.
27 Pediatric Trauma Assessment
Trauma is the leading cause of death in children >1 year old. Key anatomic and physiologic differences from adults that affect trauma management:
| Feature | Pediatric Difference | Clinical Implication |
|---|---|---|
| Head | Proportionally larger, thinner calvarium | Higher incidence of head injury; more vulnerable to intracranial hemorrhage |
| Skeleton | More cartilaginous, more compliant | Internal organ injury without fractures (ribs protect less); Waddell triad (bumper hits femur + chest/abdomen + head hits ground) |
| Blood volume | 80 mL/kg (vs 70 mL/kg adult) | Tachycardia is the earliest sign of shock (hypotension is a LATE sign — children compensate until ~25-30% blood loss) |
| Airway | Large tongue, anterior larynx, cricoid narrowing | Use age-appropriate equipment; uncuffed ETT for younger children |
| C-spine | Higher fulcrum (C2-C3 in children vs C5-C6 in adults); ligamentous laxity | SCIWORA (spinal cord injury without radiographic abnormality); pseudosubluxation at C2-C3 |
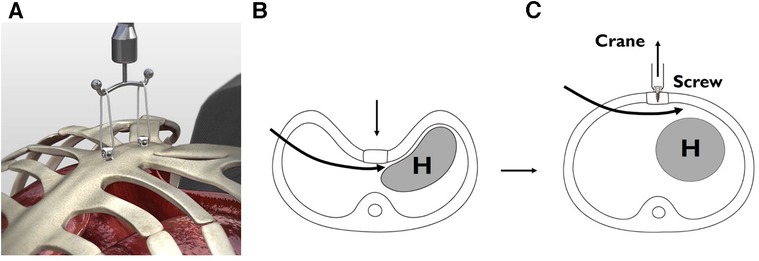
Weight-Based Calculations
Estimated weight formulas: Age 1-10 years: (age x 2) + 10 = weight in kg. The Broselow tape (length-based resuscitation tape) provides weight estimation and corresponding drug doses, equipment sizes, and fluid volumes by measuring the child's length — essential for resuscitation of children when actual weight is unknown.
Pediatric Fluid Resuscitation
Initial bolus: 20 mL/kg isotonic crystalloid (NS or LR). Reassess after each bolus. If no improvement after 2 boluses (40 mL/kg), transfuse PRBCs at 10 mL/kg. Activate massive transfusion protocol if ongoing hemorrhage. Blood volume calculation for estimated allowable blood loss: EBV = weight (kg) x blood volume per kg (80 mL/kg for infants, 70 mL/kg for children).
28 Solid Organ Injury, Non-Accidental Trauma & Burns
Nonoperative Management of Solid Organ Injuries
Children have a higher success rate of nonoperative management (NOM) of solid organ injuries than adults — >95% of splenic injuries and >90% of liver injuries in hemodynamically stable children are managed nonoperatively. This higher NOM success rate is attributed to more robust splenic capsule, stronger vasoconstrictive response, and generally fewer comorbidities.
NOM protocol: ICU admission, serial abdominal exams (q4-6h for 24-48 hours), serial hematocrit, bed rest, NPO initially. Transfusion threshold: >40 mL/kg PRBCs in 24 hours suggests NOM failure. Activity restrictions: APSA (American Pediatric Surgical Association) guidelines recommend grade + 2 weeks (e.g., Grade III spleen injury = 5 weeks restricted activity). Follow-up imaging is not routinely required for grades I-III if the child is clinically improving.
Indications for operative intervention: Hemodynamic instability not responsive to fluid resuscitation and transfusion, peritonitis (suggesting hollow viscus injury), grade V vascular injuries (shattered spleen, hepatic avulsion), and associated injuries requiring laparotomy (e.g., pancreatic duct transection, diaphragm rupture). CT with IV contrast is the imaging modality of choice for abdominal trauma in hemodynamically stable children. FAST (focused assessment with sonography in trauma) is less reliable in children than adults but useful as a screening tool.
Non-Accidental Trauma (NAT) / Child Abuse
All healthcare providers are mandated reporters. If child abuse is suspected, report to Child Protective Services (CPS) — you do not need to be certain, only to have a reasonable suspicion. Failure to report is both an ethical violation and a crime in all US jurisdictions.
Red flags for NAT:
- Injury inconsistent with developmental stage (e.g., femur fracture in a nonambulatory infant)
- Delay in seeking care, changing history, or history inconsistent with injury pattern
- Multiple injuries in various stages of healing
- Specific fracture patterns: classic metaphyseal lesions (CMLs — "bucket handle" or "corner" fractures), posterior rib fractures, spiral fractures of long bones in nonambulatory infants, skull fractures (especially bilateral or complex)
- Subdural hematomas (especially in infants — "shaken baby syndrome" / abusive head trauma)
- Retinal hemorrhages
- Burns with sharp demarcation lines, stocking/glove pattern, or on unusual locations
- Abdominal injuries in young children (duodenal hematoma, pancreatic injury — handlebar injuries are common but must distinguish from abuse)
Workup for suspected NAT: Skeletal survey (full-body X-rays — required for all children <2 years with suspected abuse; follow-up skeletal survey in 2 weeks increases sensitivity by detecting healing fractures), head CT without contrast (for children <1 year or with neurologic symptoms — MRI for further characterization), ophthalmologic exam (retinal hemorrhages), liver and pancreatic enzymes (screen for occult abdominal trauma — AST/ALT >80 should prompt abdominal CT), lipase, urinalysis, coagulation studies and CBC (to exclude bleeding disorders such as hemophilia or von Willebrand disease), and thorough documentation with photographs of all injuries with a ruler for scale.
Surgical implications: Duodenal hematoma (from a direct blow to the epigastrium — "handlebar injury" or fist) is managed nonoperatively with NGT decompression and TPN, resolving in 1-3 weeks. Pancreatic injury with ductal disruption may require distal pancreatectomy. Hollow viscus injury (jejunal perforation from shearing forces) requires operative repair. Always document injuries meticulously in the operative note for potential legal proceedings.
Burns in Children
Children have thinner skin and sustain deeper burns at lower temperatures and shorter exposure times than adults. Classification is the same as adults (superficial, partial thickness — superficial and deep, full thickness).
Modified Rule of 9s for children: The standard adult Rule of 9s overestimates the lower extremities and underestimates the head in children. The Lund-Browder chart is more accurate for pediatric BSA estimation, as it accounts for age-related proportional changes (e.g., infant head = 18% BSA vs adult 9%; infant each leg = 14% vs adult 18%).
Fluid resuscitation: The Parkland formula adapted for children: 4 mL/kg/% TBSA burned of LR over 24 hours (half in the first 8 hours from the time of injury, half over the next 16 hours), plus maintenance fluids (the 4-2-1 rule — children require maintenance fluids in addition to burn resuscitation, unlike adults). Children <20 kg should receive D5LR to prevent hypoglycemia. Target urine output: 1 mL/kg/hr (vs 0.5 mL/kg/hr in adults). Titrate to response — the Parkland formula is a starting point, not a rigid protocol.
Surgical management of burns: Escharotomy for circumferential full-thickness burns causing compartment syndrome (limbs) or respiratory compromise (chest). Early tangential excision and split-thickness skin grafting for deep partial- and full-thickness burns >20% TBSA (within 48-72 hours reduces infection and length of stay). For massive burns with limited donor sites: temporary coverage with cadaveric allograft, xenograft (porcine), or synthetic dermal substitutes (Integra). Silver sulfadiazine (topical) has been standard wound care but is increasingly replaced by mafenide acetate (better eschar penetration) or silver-impregnated dressings.
29 Key Procedures & Techniques
Laparoscopic Pediatric Surgery
Port sizes: 3 mm or 5 mm (vs 5-12 mm in adults). Insufflation pressure: 8-12 mmHg for infants and young children (vs 15 mmHg in adults). CO2 absorption is proportionally higher in children — monitor ETCO2 closely. Veress needle insertion technique modified: open (Hasson) technique preferred in small infants to avoid injury. Working space is limited — use 30-degree scopes for improved visualization.
Central Venous Access in Children
Broviac catheter: Tunneled silicone catheter placed surgically (most commonly via the internal jugular or facial vein) with the tip at the SVC-RA junction. Used for long-term IV access (TPN, chemotherapy, antibiotics). Smaller caliber than adult Hickman catheters. Port-a-Cath (implantable port): Subcutaneous reservoir connected to a tunneled catheter; accessed by percutaneous needle (Huber needle) through the skin. Preferred for intermittent access (e.g., chemotherapy cycles). Placed surgically or interventionally. Complications: line infection (most common), catheter malposition, thrombosis, mechanical failure.
Pyloromyotomy Technique (Ramstedt)
Open: RUQ transverse incision or supraumbilical semicircular incision. Deliver the pylorus through the incision. Incise the serosa longitudinally on the avascular anterior surface of the pylorus. Spread the hypertrophied muscle with a pyloric spreader (Benson) until the mucosa bulges freely. Extend the myotomy from the pyloric vein of Mayo (distal extent) to the antrum (proximal extent). Confirm mucosal integrity with air insufflation (inject air via NG tube while submerging the pylorus in saline — bubbles indicate perforation). Laparoscopic: three 3 mm ports; electrocautery to incise serosa, then spread with laparoscopic instruments.
The Ladd Procedure — Detailed Steps
(1) Midline laparotomy. (2) Eviscerate bowel. (3) Counterclockwise detorsion of volvulus. (4) Identify and divide Ladd bands crossing the duodenum. (5) Widen the mesenteric base by separating duodenum and colon. (6) Straighten the duodenum along the right abdominal gutter. (7) Place the cecum and colon in the left abdomen. (8) Appendectomy. (9) Return bowel to the abdomen in the position of nonrotation (small bowel right, colon left). Can be performed laparoscopically in stable patients.
Kasai Portoenterostomy — Technique
Right subcostal incision (extended to bilateral subcostal if needed). Mobilize the liver. Identify the fibrous biliary remnant at the porta hepatis. Dissect the remnant off the portal vein and hepatic artery. Transect the remnant at the liver surface (portal plate) — this is where the microscopic ductules are most likely to be patent. Verify bile flow (green/yellow fluid from the portal plate). Construct a 40 cm Roux-en-Y jejunal limb and anastomose it to the portal plate with interrupted absorbable sutures. An anti-reflux valve is not routinely created. Liver biopsy is performed to assess degree of fibrosis.
Pull-Through Procedures for Hirschsprung Disease
Transanal endorectal pull-through (Soave variant): (1) Submucosal dissection begins 0.5-1 cm above the dentate line, creating a circumferential mucosal sleeve. (2) The mucosal sleeve is dissected proximally (hand-sewn or cautery-assisted) to the level of the peritoneal reflection. (3) Full-thickness rectal/sigmoid colon is divided at a level confirmed to have ganglion cells on frozen section. (4) The ganglionated bowel is pulled through the aganglionic muscular cuff to the anus. (5) Coloanal anastomosis is performed at the dentate line with interrupted absorbable sutures. Can be done entirely transanally for short-segment disease or with laparoscopic assistance for longer segments. The muscular cuff is split posteriorly to prevent cuff stricture or abscess.
Circumcision
The most commonly performed surgical procedure in male neonates. Methods: Gomco clamp (bell placed over glans, foreskin drawn through the plate, clamp tightened, foreskin excised), Plastibell (ring placed between glans and foreskin, suture tied around foreskin over ring, ring falls off in 5-7 days), Mogen clamp (foreskin drawn through a slit in the clamp, excised). Complications (1-3%): bleeding (most common), infection, inadequate skin removal, excessive skin removal, meatal stenosis, urethral injury (rare but devastating — especially with Mogen clamp), concealed/buried penis. Contraindications: hypospadias (foreskin needed for repair), bleeding disorders, prematurity/illness (defer until stable).
Fundoplication (Nissen)
Performed for severe GERD refractory to medical therapy, GERD-related ALTE/BRUE, GERD with chronic aspiration pneumonia, or GERD in neurologically impaired children. Technique: laparoscopic (standard in children). Short gastric vessels are divided to mobilize the fundus. The esophageal hiatus is narrowed with crural repair (posterior). A 360-degree fundic wrap is constructed over a bougie (floppy Nissen). Gastrostomy tube is often placed simultaneously in neurologically impaired children. Complications: wrap disruption/herniation (5-15%), gas-bloat syndrome, dysphagia, dumping syndrome. Partial wraps (Thal 270-degree anterior, Toupet 270-degree posterior) may have lower dysphagia/gas-bloat rates.
30 Medications & Therapeutics
Pediatric Anesthesia Considerations
All anesthetic agents must be weight-based. Sevoflurane is the most commonly used inhalational induction agent in children (pleasant smell, rapid onset). Rapid sequence induction: succinylcholine 2 mg/kg IV (higher dose than adults due to larger volume of distribution) or rocuronium 1 mg/kg IV. Propofol: 3-5 mg/kg IV for induction (higher dose than adults). Emergence delirium is common in children aged 2-5 years after sevoflurane — treat with propofol, dexmedetomidine, or fentanyl.
Fluid & Electrolyte Replacement
| Scenario | Fluid | Rate/Dose |
|---|---|---|
| Maintenance | D5 0.45% NS + 20 mEq/L KCl | 4-2-1 rule |
| Neonatal maintenance | D10W on day 1, then D5 0.2% NS | 60-80 mL/kg/day (day 1), increasing to 150 mL/kg/day |
| Resuscitation bolus | NS or LR | 20 mL/kg, repeat x3 max |
| Transfusion | PRBCs | 10-15 mL/kg |
| Dehydration correction | NS then D5 0.45% NS | Deficit + maintenance over 24-48 hours |
Antibiotics (Weight-Based Dosing)
| Antibiotic | Dose | Common Indication |
|---|---|---|
| Ampicillin | 50-100 mg/kg/day divided q6h | NEC (with gentamicin + metronidazole) |
| Gentamicin | 4-5 mg/kg/dose q24h (neonates: 4 mg/kg/dose q24-48h) | NEC, intra-abdominal infection |
| Metronidazole | 30 mg/kg/day divided q8h | Anaerobic coverage, NEC |
| Cefoxitin | 80-160 mg/kg/day divided q6h | Perforated appendicitis |
| Piperacillin-tazobactam | 300 mg/kg/day (of piperacillin) divided q6-8h | Complicated intra-abdominal infection |
| Vancomycin | 40-60 mg/kg/day divided q6-8h | MRSA, line infections |
| Ceftriaxone | 50-100 mg/kg/day q24h | UTI, uncomplicated appendicitis |
Pain Management
Acetaminophen: 15 mg/kg PO/PR q4-6h (max 75 mg/kg/day). Neonates: 10-15 mg/kg q6-8h. IV: 15 mg/kg q6h (12.5 mg/kg for neonates). Ibuprofen: 10 mg/kg PO q6-8h (max 40 mg/kg/day; avoid <6 months). Ketorolac: 0.5 mg/kg IV q6h (max 48-72 hours; avoid in neonates, renal insufficiency). Morphine: 0.05-0.1 mg/kg IV q2-4h (caution in neonates — reduced clearance, apnea risk). Regional anesthesia: Caudal block (bupivacaine 0.25% at 1 mL/kg) — the gold standard for sub-umbilical surgery in infants and toddlers; provides 4-8 hours of analgesia. TAP (transversus abdominis plane) blocks for abdominal surgery. Epidural analgesia for major thoracic/abdominal procedures.
Chemotherapy Agents for Pediatric Tumors
| Agent | Mechanism | Common Use | Key Toxicity |
|---|---|---|---|
| Vincristine | Vinca alkaloid (microtubule inhibitor) | Wilms, RMS, neuroblastoma | Peripheral neuropathy, constipation (paralytic ileus) |
| Dactinomycin (Actinomycin D) | DNA intercalation | Wilms, RMS | Myelosuppression, radiation recall, hepatotoxicity (VOD) |
| Doxorubicin | Anthracycline (topoisomerase II inhibitor) | Hepatoblastoma, RMS, neuroblastoma | Cardiotoxicity (cumulative dose-dependent; max lifetime 450 mg/m2) |
| Cisplatin | Platinum (DNA crosslinker) | Hepatoblastoma, germ cell tumors | Nephrotoxicity, ototoxicity (requires audiometry monitoring) |
| Cyclophosphamide | Alkylating agent | RMS, Wilms (unfavorable) | Hemorrhagic cystitis (prevent with mesna), infertility |
| Etoposide | Topoisomerase II inhibitor | Germ cell tumors, neuroblastoma | Myelosuppression, secondary leukemia risk |
| Ifosfamide | Alkylating agent | RMS, germ cell tumors | Hemorrhagic cystitis, Fanconi syndrome (renal tubular toxicity) |
Prokinetics & GI Medications
Erythromycin (low dose 1-3 mg/kg IV q8h): motilin receptor agonist, used as prokinetic for feeding intolerance. Note: macrolide exposure in neonates <2 weeks old is associated with pyloric stenosis. Omeprazole: 1 mg/kg/day PO for GERD/stress ulcer prophylaxis. Ursodiol: 10-20 mg/kg/day divided BID — for TPN-associated cholestasis and post-Kasai biliary atresia. Octreotide: 1-10 mcg/kg/day IV/SC divided q8-12h — for chylothorax, chylous ascites, or high-output enterocutaneous fistula.
31 Classification Systems
Master Classification Reference Table
| System | Condition | Key Points |
|---|---|---|
| Gross classification | EA/TEF | Types A-E; Type C (EA + distal TEF) = 85% |
| Spitz classification | EA/TEF prognosis | Based on birth weight and cardiac defect |
| Bell staging (modified) | NEC | Stages I-III; pneumatosis = Stage IIA; free air = Stage IIIB |
| Todani classification | Choledochal cyst | Types I-V; Type I most common; Type V = Caroli |
| Stocker classification | CPAM/CCAM | Types 0-4; Type 1 most common (large cysts) |
| INRGSS | Neuroblastoma | L1, L2, M, MS; based on imaging risk factors |
| PRETEXT | Hepatoblastoma | I-IV based on liver sections involved |
| Altman classification | Sacrococcygeal teratoma | Types I-IV; higher type = more internal = higher malignancy risk |
| Krickenbeck classification | Anorectal malformation | Based on fistula location; replaced Wingspread |
| International VUR grading | Vesicoureteral reflux | Grades I-V on VCUG; higher = worse dilatation |
| Haller index | Pectus excavatum | Transverse/AP diameter; ≥3.25 = severe |
| CDH Study Group (A-D) | CDH defect size | A = small; D = diaphragm absent (worst prognosis) |
| Jejunoileal atresia types | Intestinal atresia | Types I-IV; IIIb = apple-peel; IV = multiple |
32 Complications & Management
| Complication | Associated Conditions | Management |
|---|---|---|
| Anastomotic stricture | EA/TEF repair, pull-through, intestinal anastomosis | Serial dilations (balloon or Savary); resection if refractory |
| Anastomotic leak | Any intestinal/esophageal anastomosis | Drainage, NPO, antibiotics; reoperation if contained leak fails |
| Adhesive small bowel obstruction | Any prior laparotomy | NGT decompression, serial exams; surgery if peritonitis or failure to resolve in 48h |
| Enterocolitis (Hirschsprung) | Hirschsprung disease (before and after pull-through) | Rectal irrigations, IV antibiotics; may require revision pull-through |
| Short bowel syndrome | NEC, volvulus, gastroschisis, multiple atresias | Intestinal rehabilitation, STEP/Bianchi, intestinal transplant |
| TPN-associated cholestasis | Prolonged TPN in neonates | Cycle TPN, reduce lipid dose, advance enteral feeds, ursodiol, Omegaven (fish oil lipid emulsion) |
| Wound infection/SSI | Any surgical procedure | Wound opening, drainage, antibiotics if cellulitis; negative pressure for complex wounds |
| Recurrent TEF | Post EA/TEF repair | Bronchoscopy to confirm, reoperation with tissue interposition (pleural flap, azygos muscle) |
| CDH recurrence | Post CDH repair (especially patch repair) | Redo repair; higher recurrence rate with patch vs primary closure |
| Dumping syndrome | Fundoplication, gastric surgery | Small frequent feeds, complex carbohydrates, acarbose; octreotide for refractory cases |
| Incisional hernia | Laparotomy in neonates | Surgical repair when symptomatic or at time of another procedure |
| Post-Kasai cholangitis | Biliary atresia post-Kasai | IV antibiotics (third-generation cephalosporin + aminoglycoside); prophylactic TMP-SMX |
33 Medications Master Table
| Drug | Class | Pediatric Dose | Key Notes |
|---|---|---|---|
| Acetaminophen | Analgesic/antipyretic | 15 mg/kg PO/PR q4-6h; 12.5-15 mg/kg IV q6h | Max 75 mg/kg/day; hepatotoxicity in overdose |
| Ibuprofen | NSAID | 10 mg/kg PO q6-8h | Avoid <6 months; renal/GI risks |
| Morphine | Opioid | 0.05-0.1 mg/kg IV q2-4h | Reduce dose in neonates; apnea monitoring |
| Ketorolac | NSAID (IV) | 0.5 mg/kg IV q6h (max 72h) | Excellent adjunct; avoid in renal failure/neonates |
| Ampicillin | Penicillin | 50-100 mg/kg/day divided q6h | NEC regimen, neonatal sepsis |
| Gentamicin | Aminoglycoside | 4-5 mg/kg q24h | Monitor levels (peak/trough); nephro/ototoxicity |
| Metronidazole | Nitroimidazole | 30 mg/kg/day divided q8h | Anaerobic coverage |
| Piperacillin-tazobactam | Extended-spectrum penicillin | 300 mg/kg/day divided q6-8h | Broad-spectrum; complicated infections |
| Vancomycin | Glycopeptide | 40-60 mg/kg/day divided q6-8h | MRSA coverage; monitor trough levels |
| Ceftriaxone | 3rd-gen cephalosporin | 50-100 mg/kg/day q24h | Avoid in neonates (bilirubin displacement) |
| Omeprazole | PPI | 1 mg/kg/day PO | GERD, stress ulcer prophylaxis |
| Ursodiol | Bile acid | 10-20 mg/kg/day divided BID | TPN cholestasis, post-Kasai |
| Octreotide | Somatostatin analogue | 1-10 mcg/kg/day divided q8-12h | Chylothorax, high-output fistula |
| Erythromycin | Macrolide/prokinetic | 1-3 mg/kg IV q8h (prokinetic dose) | Motilin receptor agonist; pyloric stenosis risk in neonates |
| Vincristine | Vinca alkaloid | 0.05 mg/kg IV (max 2 mg) | Neuropathy; constipation; vesicant |
| Dactinomycin | Antitumor antibiotic | 0.045 mg/kg IV (max 2.5 mg/cycle) | Hepatotoxicity (VOD); radiation recall |
| Doxorubicin | Anthracycline | 30-75 mg/m2 IV per cycle | Cumulative cardiotoxicity (max 450 mg/m2 lifetime) |
| Cisplatin | Platinum agent | 80-100 mg/m2 IV per cycle | Nephrotoxicity (aggressive hydration); ototoxicity |
| Cyclophosphamide | Alkylating agent | 1.2-2.2 g/m2 IV per cycle | Hemorrhagic cystitis (mesna); infertility |
| Teduglutide | GLP-2 analogue | 0.05 mg/kg SC daily | SBS intestinal adaptation; monitor for polyps |
34 Abbreviations Master List
| Medication | Dose | Route |
|---|---|---|
| Epinephrine | 0.01 mg/kg (0.1 mL/kg of 1:10,000) | IV/IO |
| Atropine | 0.02 mg/kg (min 0.1 mg) | IV/IO |
| Amiodarone | 5 mg/kg | IV/IO |
| Adenosine | 0.1 mg/kg (max 6 mg), then 0.2 mg/kg (max 12 mg) | Rapid IV push |
| Succinylcholine | 2 mg/kg (infant), 1-1.5 mg/kg (child) | IV |
| Rocuronium | 1 mg/kg (RSI) | IV |
| Dextrose (D10W) | 2-4 mL/kg | IV (for hypoglycemia) |
| Calcium gluconate 10% | 0.5-1 mL/kg (max 20 mL) | Slow IV push |
| NS bolus | 20 mL/kg | IV (repeat x3 max) |
| PRBCs | 10-15 mL/kg | IV |
| Abbreviation | Meaning |
|---|---|
| ARM | Anorectal malformation |
| BA | Biliary atresia |
| BPD | Bronchopulmonary dysplasia |
| BPS | Bronchopulmonary sequestration |
| BSA | Body surface area |
| CDH | Congenital diaphragmatic hernia |
| CF | Cystic fibrosis |
| CLE | Congenital lobar emphysema |
| COG | Children's Oncology Group |
| CPAM | Congenital pulmonary airway malformation |
| EA | Esophageal atresia |
| ECMO | Extracorporeal membrane oxygenation |
| ETT | Endotracheal tube |
| HAEC | Hirschsprung-associated enterocolitis |
| HFOV | High-frequency oscillatory ventilation |
| HPS | Hypertrophic pyloric stenosis |
| INRGSS | International Neuroblastoma Risk Group Staging System |
| IVH | Intraventricular hemorrhage |
| LHR | Lung-to-head ratio |
| MIS | Minimally invasive surgery |
| NAT | Non-accidental trauma |
| NEC | Necrotizing enterocolitis |
| NOM | Nonoperative management |
| NWTS | National Wilms Tumor Study |
| OI | Oxygenation index |
| PAS | Pediatric Appendicitis Score |
| PDA | Patent ductus arteriosus |
| PPD | Primary peritoneal drain |
| PPHN | Persistent pulmonary hypertension of the newborn |
| PPV | Patent processus vaginalis |
| PRETEXT | PRE-Treatment EXTent of disease |
| PSARP | Posterior sagittal anorectoplasty |
| PUV | Posterior urethral valves |
| RMS | Rhabdomyosarcoma |
| ROP | Retinopathy of prematurity |
| SBS | Short bowel syndrome |
| SCT | Sacrococcygeal teratoma |
| SIOP | Societe Internationale d'Oncologie Pediatrique |
| STEP | Serial transverse enteroplasty |
| TBSA | Total body surface area |
| TEF | Tracheoesophageal fistula |
| TPN | Total parenteral nutrition |
| VACTERL | Vertebral, Anorectal, Cardiac, Tracheoesophageal, Renal, Limb |
| VCUG | Voiding cystourethrogram |
| VMA/HVA | Vanillylmandelic acid / Homovanillic acid |
| VUR | Vesicoureteral reflux |
References & Figure Sources
Landmark Scoring Systems & Quick Reference
| Condition | Key Diagnostic Finding | Key Treatment |
|---|---|---|
| EA/TEF | NG tube coils in upper pouch on CXR | Thoracotomy, fistula ligation, esophageal anastomosis |
| CDH | Bowel in chest on CXR, scaphoid abdomen | Stabilize first, delayed repair |
| Duodenal atresia | "Double bubble" on AXR | Duodenoduodenostomy (diamond) |
| Malrotation/volvulus | Bilious emesis + abnormal UGI | Ladd procedure (EMERGENCY) |
| Meconium ileus | Microcolon on contrast enema; CF history | Gastrografin enema; surgery if complicated |
| Pyloric stenosis | Olive mass; US ≥3 mm thickness, ≥15 mm length | Correct alkalosis FIRST, then Ramstedt pyloromyotomy |
| Intussusception | Target sign on US | Air enema reduction; surgery if fails |
| NEC | Pneumatosis on AXR | NPO, antibiotics; surgery for free air or deterioration |
| Hirschsprung | Failed meconium passage; biopsy aganglionosis | Pull-through (Soave/Duhamel/Swenson) |
| Biliary atresia | Conjugated hyperbilirubinemia >2 weeks | Kasai portoenterostomy before 60 days |
| Neuroblastoma | Elevated urine VMA/HVA; mass crossing midline | Risk-stratified: surgery ± chemo ± immunotherapy |
| Wilms tumor | Abdominal mass NOT crossing midline | Nephrectomy + chemo (VCR/DACT ± DOX) |
| Testicular torsion | Absent cremasteric reflex; acute scrotal pain | Emergent bilateral orchiopexy |
| Undescended testis | Nonpalpable or inguinal testis | Orchiopexy at 6-12 months |
Key Trials & Guidelines
- Spitz L, Kiely EM, Morecroft JA, Drake DP. Oesophageal atresia: at-risk groups for the 1990s. J Pediatr Surg. 1994;29(6):723-725. PMID: 8078005
- Lally KP, Lasky RE, Lally PA, et al. Standardized reporting for congenital diaphragmatic hernia — an international consensus. J Pediatr Surg. 2013;48(12):2408-2415. PMID: 24314178
- Pena A, Devries PA. Posterior sagittal anorectoplasty: important technical considerations and new applications. J Pediatr Surg. 1982;17(6):796-811. PMID: 6761417
- De la Torre-Mondragon L, Ortega-Salgado JA. Transanal endorectal pull-through for Hirschsprung's disease. J Pediatr Surg. 1998;33(8):1283-1286. PMID: 9722005
- Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369(9579):2106-2120. PMID: 17586306
- Dome JS, Graf N, Geller JI, et al. Advances in Wilms tumor treatment and biology: progress through international collaboration. J Clin Oncol. 2015;33(27):2999-3007. PMID: 26304889
- Coran AG, Adzick NS, Krummel TM, et al. Pediatric Surgery. 7th ed. Elsevier; 2012. (Standard pediatric surgery reference)
- Holcomb GW III, Murphy JP, St Peter SD. Holcomb and Ashcraft's Pediatric Surgery. 7th ed. Elsevier; 2020.
- Bell MJ, Ternberg JL, Feigin RD, et al. Neonatal necrotizing enterocolitis: therapeutic decisions based upon clinical staging. Ann Surg. 1978;187(1):1-7. PMID: 413500
- Snodgrass W. Tubularized, incised plate urethroplasty for distal hypospadias. J Urol. 1994;151(2):464-465. PMID: 8283561
- Kim HB, Fauza D, Garza J, et al. Serial transverse enteroplasty (STEP): a novel bowel lengthening procedure. J Pediatr Surg. 2003;38(3):425-429. PMID: 12632361
- Kasai M, Suzuki S. A new operation for "non-correctable" biliary atresia — hepatic portoenterostomy. Shujutsu. 1959;13:733-739.
- Peters CA, Skoog SJ, Arant BS Jr, et al. Summary of the AUA guideline on management of primary vesicoureteral reflux in children. J Urol. 2010;184(3):1134-1144. PMID: 20650499
Textbooks & Reference Works
- Coran AG, Adzick NS, Krummel TM, Laberge JM, Shamberger RC, Caldamone AA. Pediatric Surgery. 7th ed. Elsevier; 2012.
- Holcomb GW III, Murphy JP, St Peter SD. Holcomb and Ashcraft's Pediatric Surgery. 7th ed. Elsevier; 2020.
- Puri P, Hollwarth ME. Pediatric Surgery: Diagnosis and Management. 2nd ed. Springer; 2019.
- Grosfeld JL, O'Neill JA Jr, Fonkalsrud EW, Coran AG. Pediatric Surgery. 6th ed. Mosby; 2006.
- Oldham KT, Colombani PM, Foglia RP, Skinner MA. Principles and Practice of Pediatric Surgery. Lippincott Williams & Wilkins; 2005.